
超高效合相色谱-串联质谱法测定中药材中10种偶氮染料
2021-12-02 09:23杨光勇郭金喜沙丽娜
质谱学报 2021年6期
杨光勇,薛 光,郭金喜,沙丽娜
(1.新疆维吾尔自治区产品质量监督检验研究院,新疆 乌鲁木齐 830011;2.新疆维吾尔自治区药品检验研究院,新疆 乌鲁木齐 830054)
偶氮染料(ADs)是石油、建材等行业常用的工业染色剂,具有染色效果好、不易脱色、成本低廉等优势,其代谢产物芳香胺具有强烈的致癌性[1-2]。ADs进入动物体内可在动物肝脏中被还原成芳香胺,芳香胺分子上的氨基具有代谢活性,可进一步形成羟胺,从而破坏蛋白质和DNA结构,使细胞发生变异[3],因此,ADs被多个国家和地区明令禁止用于食物染色。相关研究[4-5]显示,我国ADs的污染状况不容乐观,一些不法之徒为使产品更加美观,将工业染色剂非法添加至中药材中,甚至将药渣表面喷涂ADs后再次出售,严重扰乱了正常的市场经济秩序。随着国内消费者对健康、养生的日益重视,购买安全、放心的中药材及中药饮片已成为人们提高自身免疫力、加强身体素质的需求。同时,传统中医药在防治非典型肺炎、新冠肺炎等重大传染疾病时发挥了重要作用,未来将会服务国内外更多人群。因此,建立中药材中多种ADs的测定方法,对于从源头控制产品质量、保障消费者食药安全、保护中国特色医药等具有重要意义。
ADs的检测方法有薄层色谱法[6]、分光光度法[7]、电化学分析法[8]、液相色谱法[9-10]等,这些方法存在定量能力有限、灵敏度低、选择性差等问题。近年来,研究者采用气相色谱-质谱法(GC-MS)[11]、液相色谱-串联质谱法(LC-MS/MS)[12-14]、毛细管电泳-串联质谱法(CE-MS/MS)[15]等检测ADs。其中,GC-MS法需要对样品进行衍生化处理,操作繁冗复杂,测定时间长,难以实现高通量分析;CE-MS/MS法在方法精密度、再现性等方面存在不足,限制了其进一步应用和推广。还有研究采用核磁共振波谱法[16]、红外光谱法[17]、高分辨质谱直接测定法[18]等快速筛查样品中的ADs,但这些方法均无法实现准确定量。
超高效合相色谱(ultra performance convergence chromatography, UPC2)是以超临界CO2为主要流动相的色谱系统,兼具超临界流体色谱的优势和传统高效液相色谱的易用性,已成功应用于双酚A[19]、对羟基苯甲酸酯[20]等脂溶性化合物的分析。本实验拟采用固相萃取法(SPE)净化、浓缩,超高效合相色谱-三重四极杆质谱法(UPC2-MS/MS)测定中药材中10种ADs,旨在为测定中药材中脂溶性工业染料提供思路。
1 实验部分
1.1 主要仪器与装置
UPC2-TQS超高效合相色谱-三重四极杆质谱仪:美国Waters公司产品,配有电喷雾电离源(ESI)和MassLynx V4.1数据处理软件;GCMS-QP2020气相色谱-质谱仪:日本Shimadzu公司产品,配有电子轰击源(EI)、GCMS Solution V4.2数据处理软件及NIST 2014数据库;Syncore Analyst平行蒸发定量浓缩仪:瑞士Buchi公司产品;2-16P离心机:德国Sigma公司产品;S50R超声波清洗器:德国Elma公司产品。
1.2 主要材料与试剂
苏丹红专用SPE柱、中性氧化铝SPE柱:规格均为500 mg/6 mL,北京迪科马科技有限公司产品;苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、苏丹红G、苏丹红7B、苏丹橙G、苏丹黄、对位红、苏丹红B标准品:纯度≥97.0%,德国Dr.Ehrenstorfer公司产品;甲酸、甲酸铵、正己烷、甲醇、丙酮:色谱纯,美国Thermo Fisher公司产品。
成品药材红枣、枸杞、红花、朱砂、红藤、丹参:购自本地市场。
1.3 标准溶液的配制
精密称取适量的各标准品,用正己烷制成0.1 g/L单标储备液,准确移取适量的各储备液,用正己烷稀释成100 μg/L混合标准使用液。
1.4 样品前处理
将红枣、枸杞样品置于-80 ℃冰箱中冷冻30 min,取出后立即放入粉碎机中,并加入适量的干冰中和摩擦热,粉碎后过50目筛;其他种类药材样品直接粉碎后过50目筛。称取2.00 g样品于15 mL具塞玻璃离心管中,加入10 mL正己烷,超声提取10 min,以5 000 r/min离心5 min,移取正己烷层,残渣用正己烷重复提取2次,每次10 mL;合并提取液,转移至经5 mL正己烷活化后的中性氧化铝SPE柱,以5 mL正己烷淋洗,弃去全部流出液,以6 mL含30%丙酮的正己烷溶液洗脱,收集洗脱液并定量浓缩至1 mL,过0.22 μm滤膜。
1.5 实验条件
1.5.1GC-MS条件 色谱柱:DB-5MS石英毛细管柱(30 m×0.25 mm×0.25 μm);升温程序:100 ℃保持2 min,以5 ℃/min升温至250 ℃,保持5 min;进样口温度280 ℃;载气(He)流速1.0 mL/min;进样量1 μL,不分流进样;电子轰击能量70 eV,传输线温度250 ℃,离子源温度220 ℃;质量扫描范围m/z50~800;溶剂延迟3 min。
1.5.2UPC2-MS/MS条件 色谱柱:Viridis HSS C18 SB柱(150 mm×2.1 mm×1.8 μm),柱温40 ℃;流动相:超临界CO2-10 mmol/L甲酸铵甲醇溶液(88∶12,V/V);流动相流速1.5 mL/min;系统背压13.79 MPa;补偿液:97%甲醇水溶液(含0.2%甲酸),流速0.3 mL/min,进样体积5 μL;电喷雾电离源正离子模式;离子源温度150 ℃;毛细管电压2.5 kV;去溶剂气流速750 L/h;去溶剂气温度400 ℃;多反应监测模式(MRM)采集。10种ADs的基本信息列于表1。
2 结果与讨论
2.1 样品提取及净化条件的优化
偶氮染料极性较弱,可使用丙酮、正己烷、乙醚等有机溶剂进行提取。为减少样品提取液中的极性干扰物并使提取更充分,实验选用30 mL正己烷分3次提取。通过比较振荡、超声两种提取方式,以及在5、10、15 min不同超声时间下加标样品(25 μg/kg)的回收率可知,超声提取更加快速高效,最佳提取时间为10 min,结果示于图1。经过3次重复提取后,加标样品的回收率即可满足一般化学分析的要求。

表1 10种ADs的保留时间、相对分子质量、母离子和子离子Table 1 Retention time, relative molecular mass (Mr), parent ions, daughter ions of ten kinds of ADs
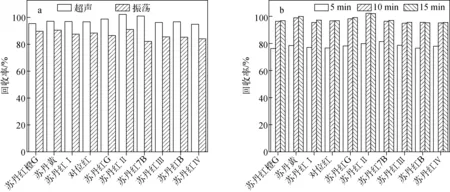
图1 加标水平为25 μg/kg的红花样品在不同提取方式(a)、不同超声时间(b)下10种ADs回收率Fig.1 Recoveries of ten kinds of ADs spiked in safflower at 25 μg/kg with different extraction ways (a) and different ultrasonic extraction time (b)
样品提取液中含有较多杂质,可能对测定产生干扰,并造成仪器污染。通过采用GC-MS扫描各样品提取液,再利用NIST 2014数据库对扫描所得的总离子流图中各色谱峰进行检索,发现提取液中存在大量的长链脂肪烃、萜类及其含氧衍生物、游离脂肪酸,这些化合物的峰面积之和约占色谱峰总面积的82%~95%,部分化合物的响应高达108。因此,实验采用SPE法对加标样品(25 μg/kg)的提取液进行净化,并比较了不同SPE柱的净化效果。按1.4节方法,分别用中性氧化铝SPE柱和苏丹红专用SPE柱净化提取液,并分析回收率数据及待测溶液的GC-MS图谱。结果表明,经净化后,提取液GC-MS图谱中杂质峰簇明显减少,各ADs回收率均有所提高,示于图2。这可能是因为净化过程有效去除了提取液中大部分易引起离子化抑制效应的样品基质;使用2种SPE柱时,各ADs的回收率没有明显差别,但中性

图2 净化前、后红花样品(加标水平为25 μg/kg)提取液的GC-MS总离子流色谱图(a)和回收率(b)Fig.2 TIC (a) and recoveries (b) of the extract of safflower (the spiked level of 25 μg/kg) before and after purification
氧化铝SPE柱价格低廉,且可通过自行填装进一步降低成本。因此,实验选用中性氧化铝SPE柱作为前处理净化柱。
2.2 色谱和质谱条件优化
2.2.1色谱柱的选择 由于各ADs的结构和极性极为相似,部分ADs的母离子及子离子的质荷比均相同,不容易实现基线分离,进而影响定性和定量的准确性,这为选择色谱柱带来一定难度。本实验在流动相为超临界CO2-甲醇(95∶5,V/V)、流速1.0 mL/min、柱温45 ℃、背压13.79 MPa的色谱条件下,考察了采用Viridis HSS C18 SB(150 mm×2.1 mm×1.8 μm)、Torus 1-AA(100 mm×3.0 mm×1.8 μm)2种UPC2色谱柱测定ADs混合标准溶液。结果表明,选用1-AA色谱柱无法有效分离苏丹红Ⅰ/苏丹红Ⅱ、苏丹红B/苏丹红Ⅳ这2组化合物,这可能是该色谱柱中以π-π、偶极-偶极为主要作用力的色谱填料无法识别ADs分子苯环上甲基的微小变化;选用HSS C18 SB色谱柱时,各ADs分离效果良好(分离度≥3.64),但苏丹红G色谱峰略有拖尾,峰形不佳。综上,实验选择HSS C18 SB色谱柱作为分析柱继续优化色谱条件。
2.2.2色谱柱温度和系统背压的选择 系统背压和色谱柱温度的变化会影响超临界CO2的密度,从而改变其洗脱能力。当背压增大、柱温降低时,流动相密度增大,化合物保留时间缩短。实验分别考察了不同柱温(20、30、40、50 ℃)、不同背压(12.07、13.10、13.79、14.48 MPa)对分离效果的影响。结果表明,各ADs分离效果并未随背压的改变而发生明显变化;柱温升高时,各ADs保留时间延长,分离趋势增加;当柱温超过40 ℃时,保留时间反而缩短,可能因为此时柱温对分子能量的影响超过了其对流动相洗脱能力的影响。综合考虑分离度、系统压力极限等因素,选取柱温40 ℃、背压13.79 MPa为测定条件。
2.2.3共溶剂的选择 在UPC2中引入适量共溶剂可有效改善超临界CO2的选择性和洗脱能力。甲醇、乙腈、异丙醇等均是UPC2常用的共溶剂,通常选用洗脱强度值较高的共溶剂可获得更优异的色谱峰形。为获得更尖锐、对称的色谱峰形,实验选用含10 mmol/L甲酸铵的甲醇溶液作为共溶剂。结果表明,在共溶剂中加入适量甲酸铵,可降低ADs分子上各官能团与未键合硅醇羟基的相互作用,有效改善了拖尾峰的色谱峰形。在上述优化的色谱条件下,进一步调整流动相组成及流速,以缩短分析时间,测定的10种ADs混合标准溶液(50 μg/L)的提取离子流色谱图示于图3。
2.2.4补偿液组成及流速的优化 柱塞泵通过三通组件将补偿液输送至色谱柱后端,补偿液再通过另一个三通组件将流出色谱柱的化合物携带至质谱仪。补偿液不参与色谱分离,但其流速和组成对化合物色谱峰形、离子化效率等均有较大影响。ADs分子结构中含N=N键,有利于氢加合物离子的形成。因此,实验选用含0.2%甲酸的甲醇溶液作为补偿液,并进一步考察了补偿液在不同流速(0.1、0.2、0.3、0.4 mL/min)下对测定结果的影响。结果表明,补偿液流速较低时,各化合物质谱响应较弱,色谱峰半峰宽较大;当流速为0.3 mL/min时,各ADs色谱峰形和响应即可达到最佳状态,继续增大流速反而会使质谱响应减弱,可能因为此时系统压力过大,迫使背压阀开启排放功能,分流了部分目标化合物。

注:峰序号与表1各化合物序号一致图3 10种ADs混合标准溶液(50 μg/L)的提取离子流色谱图Fig.3 Extractedion current chromatogram of ten kinds of ADs mixed standard solution
此外,实验发现在补偿液中加入适量纯水可进一步提高ADs的质谱响应。通过比较加入不同纯水含量 (2%、3%、4%、5%、6%)的补偿液时ADs的响应强度可知,纯水的最佳含量为3%。当纯水含量超过5%后,系统压力波动超过仪器警告阈值,设备始终无法就绪,可能因为纯水的高比热容使系统排放阀无法正常控温,从而影响了超临界状态的稳定性。
2.2.5质谱采集参数的优化 将各标准储备液适当稀释后,依次注入三重四极杆质谱仪,开启IntelliStart自动调谐功能,采用仪器自动优化所得的锥孔电压、碰撞能量、驻留时间等参数。
2.3 方法学验证
2.3.1基质效应 分别配制系列基质匹配标准溶液和系列空白溶剂标准溶液,按基质效应(ME)=|(基质匹配标准曲线斜率/空白溶剂标准曲线斜率-1)|×100%进行考察。ME值≤20%为弱基质效应;ME值处于20%~50%为中等基质效应;ME值>50%为强基质效应。结果表明,各样品的ME值≤13.4%,基质效应较弱。
2.3.2线性范围、检出限和定量限 用正己烷配制各组分质量浓度均为1.0、2.5、5.0、10、25、50、100 μg/L的系列混合标准溶液,以对应质量浓度为横坐标,目标物定量离子峰面积为纵坐标绘制标准曲线。结果表明,10种ADs在各自的浓度范围内线性关系良好,相关系数(r2)≥0.998 8,列于表2。
在阴性红花样品中加入适量各标准品,分别以信噪比(S/N)为3和10计算检出限(LOD)和定量限(LOQ),结果列于表2。10种ADs的LOD和LOQ分别为0.2~1.0 μg/kg、0.5~2.5 μg/kg。
2.3.3回收率与精密度 在阴性红花样品中加入适量各标准品,制成各ADs含量均为2.5、5、25 μg/kg的加标样品,每个加标水平制备3份,按1.4节方法处理后测定,得到样品的平均回收率为92.0%~106.2%(n=9),相对标准偏差为1.9%~4.2%(n=9),结果列于表2。
2.4 实际样品的测定
按照建立的方法对红枣、枸杞、红花、朱砂、红藤、丹参等6种共25份样品进行检测,其中1份红花样品中检出苏丹红Ⅰ和苏丹红Ⅳ,含量合计7.2 mg/kg,其他样品均未检出这10种偶氮染料。为进一步确证测定结果,实验对阳性样品进行了二级质谱扫描,示于图4。保留时间为1.017 min的化合物其主要子离子分别为m/z93.1、156.2、231.2,推测其母离子裂解机理示于图4a,其中子离子m/z231.2是由母离子脱去H2O后形成的,该子离子活性较高,可通过重排、开环等反应进一步碎裂成m/z142.1、193.1、217.1碎片离子。保留时间为4.230 min的化合物,其主要子离子为m/z90.9、156.3、224.4、260.5、276.2、363.3、366.1,推测其母离子裂解机理示于图4b,母离子脱去H2O后形成m/z363.3碎片离子,可在反应池中进一步碎裂成m/z142.8、213.9、196.0等碎片离子。根据以上二级质谱图数据,可排除样品假阳性的可能性。

表2 10种ADs的线性范围、相关系数、检出限、定量限、回收率及相对标准偏差Table 2 Linear range, correlation coefficient (r2), LOD, LOQ, recovery and RSD of ten kinds of ADs

图4 红花样品中苏丹红Ⅰ(a)、苏丹红Ⅳ(b)的子离子扫描图Fig.4 Product ion scan spectra of Sudan Ⅰ(a) and Sudan Ⅳ(b) in the safflower
3 结论
本文建立了UPC2-MS/MS法测定中药材中多种偶氮染料含量,采用优化的样品前处理方式,有效减小了基质干扰,提高了检测灵敏度。该方法快速准确、灵敏度高、专属性好、绿色环保,可在4.5 min内实现10种偶氮染料的分离和测定,适用于实际样品的检测。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15
应用化工(2021年4期)2021-05-20
食品安全导刊(2020年14期)2020-12-04
化工管理(2020年26期)2020-10-09
中国粮油学报(2019年4期)2019-07-12
山东化工(2019年2期)2019-02-21
现代食品(2018年16期)2018-11-02
中成药(2018年2期)2018-05-09
健康博览(2017年12期)2018-02-06
中成药(2017年4期)2017-05-17
