
基于HPLC-UV-ELSD指纹图谱法同时测定绞股蓝中4种成分及不同产地药材质量评价*
2021-11-21 08:17王源源
中医药导报 2021年5期
王源源,丁 鹏
(北京中医药大学东直门医院,北京 100700)
绞股蓝为多年生草质藤本植物,始载于《救荒本草》,又名七叶胆、福音草、遍地生根、天堂草、五叶参、七叶参、超人参等[1-2]。据《香港中药材标准》(第5期)记载,绞股蓝来源于葫芦科植物绞股蓝Gynostemma pentaphyllum(Thunb.)Makino的干燥地上部分。生长于山谷密林(海拔100~3 200 m)、山坡疏林及灌丛中,在陕西、甘肃和长江以南各地广泛分布[3]。有清热解毒、益气健脾、养阴生津、化痰止咳、养心安神之功效,用于治疗体虚乏力、虚劳失精、白细胞减少、高脂血、病毒性肝炎、慢性胃肠炎、慢性气管炎等病症,也比较适合肥胖和长期疲劳的人群,近年来被冠以“南方人参”和“第二人参”的美誉[4-6]。绞股蓝药材中含有绞股蓝皂苷、黄酮类、绞股蓝糖苷、微量元素、维生素、矿物质等多种化学成分,目前普遍认为达玛烷型四环三萜皂苷和黄酮是其两大主要有效成分[7-10]。现代药理研究表明[11-13],以绞股蓝皂苷A、绞股蓝皂苷XLIX为代表的皂苷类成分和以芦丁和槲皮素为代表的黄酮类成分均具有抑制肿瘤生长、降低血糖、保护心脑血管系统、减少氧化应激、调节血脂平衡等作用。
近年来,国内学者主要对绞股蓝药材中黄酮类或皂苷类成分分别进行指纹图谱、化学成分或药理作用的研究,但仅以其中单一成分对绞股蓝的质量进行评价,难以体现中药多成分、多靶标协同作用的整体性,而相关研究报道鲜见。《中华人民共和国药典》(2015版)未收载绞股蓝药材相关标准,部分地方中药材标准对其质量的全面控制还不够完善,而药材的质量易受多方面因素(如品种、产地、气候、生态环境、不同批次及不同产地等)的影响。故本试验将通过HPLC-UVELSD串联指纹图谱和定量分析法综合评价不同产地绞股蓝药材的质量,以期能整体上控制其质量,为绞股蓝药材临床使用的有效性和稳定性提供实验支持。
1 实验材料
1.1 仪器Agilent1200型高效液相色谱仪(美国安捷伦公司),包括UV检测器,四元梯度泵,软件Empower Pro色谱工作站;Alltech3300型蒸发光散射检测器(ELSD)(格雷斯中国有限公司);Milli Pore Advantage A10型自动纯水机(美国密理博公司);KQ5200 DE型数控超声波清洗器(昆山市超声仪器有限公司);XS205DU型电子分析天平(瑞士梅勒特有限公司)。
1.2 试药 对照品包括芦丁(批号:100080-201610,纯度≥91.9%)、槲皮素(批号:100081-201408,纯度≥99.1%)、绞股蓝皂苷A(批号:157752-01-7,纯度≥98.0%)、绞股蓝皂苷XLIX(批号:94987-08-3,纯度≥98.0%)均购于中国食品药品检定研究院;流动相乙腈和甲醇(色谱级);蒸馏水(屈臣氏)。其余试剂为分析纯。不同产地绞股蓝Gynostemma pentaphyllum(Thunb.)Makino药材共15批均购买药材市场或药店,见表1。

表1 15批绞股蓝药材来源信息
1.3 观察指标与方法 观察指标:(1)HPLC-UV-ELSD研究绞股蓝药材指纹图谱:研究15批不同产地绞股蓝药材的共有模式;(2)测定绞股蓝药材中槲皮素、芦丁、绞股蓝皂苷A和绞股蓝皂苷XLIX 4种成分的含量。检测方法:(1)HPLC-UV-ELSD指纹图谱研究:色谱条件为Thermo BDS C18色谱柱(4.6 mm×250 mm,5 μm);乙腈(A)-0.5%乙酸水(B)溶液为流动相;梯度洗脱程序为:0~10 min,10%(A),10~18 min,10%→20%(A),18~40 min,20%→45%(A),40~55 min,45%→60%(A),55~70 min,60%→100%(A);检测波长为360 nm;柱温为30℃;ELSD漂移管温度为80℃,载气流量为1.5 L/min;进样量为10 μL,流速为1.0 mL/min。分别精密称取芦丁、槲皮素、绞股蓝皂苷A和绞股蓝皂苷XLIX适量对照品,置于5 mL的棕色量瓶中,缓缓滴加甲醇溶液至刻度定容溶解,制备的混合对照品溶液终浓度分别为芦丁0.256 mg/mL、槲皮素0.131 mg/mL、绞股蓝皂苷A 0.142 mg/mL、绞股蓝皂苷XLIX 0.185 mg/mL。然后,称取绞股蓝药材(过65目筛)约2.0 g,精密称重后,置索氏提取器中,加石油醚(60~90℃)适量,加热回流提取至无色,冷却,弃去石油醚液,挥干溶剂。再次加入甲醇80 mL,进行4 h的回流提取,冷却后加甲醇定容稀释至100 mL,摇匀,所有样品进样前需过0.45 μm的微孔滤膜。(2)含量测定:在指纹图谱研究的基础上同时定量分析4种成分的含量,精密吸取各对照品母液,稀释配制一系列浓度的混合对照品溶液,按已定的高效液相色谱条件进样分析,对照品峰面积(A)为纵坐标(Y),进样量(ng)为横坐标(X),根据建立的标准曲线,计算得出各指标性成分的线性回归方程。
2 结 果
2.1 HPLC-UV-ELSD指纹图谱研究结果
2.1.1 精密度试验 取重复性试验中的同一供试品溶液连续6次进样,参照峰为4号(芦丁)峰,分别计算11个共有峰的相对保留时间RSD和相对峰面积RSD。结果显示,各个共有峰的相对保留时间与相对峰面积的RSD均小于2.0%,并且各共有指纹峰的相似度均大于0.990,表明高效液相色谱仪的精密度良好。
2.1.2 稳定性试验 取重复性试验中的同一供试品溶液,分别于0、2、4、8、12、24 h进样分析,参照峰为4号(芦丁)峰,分别计算11个共有峰的相对保留时间RSD和相对峰面积RSD。结果显示,各个共有峰的相对保留时间与相对峰面积的RSD均小于2.0%,并且各共有指纹峰的相似度均大于0.990,表明本次实验制备的供试品溶液在24 h内较稳定。
2.1.3 重复性试验 精密称取6份过65目筛的绞股蓝样品(S1)2.0 g,样品溶液按照已定的方法制备,过滤进样分析,参照峰为4号(芦丁)峰,分别计算11个共有峰的相对保留时间RSD和相对峰面积RSD。结果显示,各个共有峰的相对保留时间与相对峰面积的RSD均小于2.0%,并且各共有指纹峰的相似度均大于0.990,表明本次实验方法的重复性良好。
2.1.4 HPLC-UV-ELSD指纹图谱的建立及其相似度分析 分别精密吸取10 μL的对照品溶液及样品溶液,进样分析,记录15批不同来源绞股蓝药材在70 min内的HPLC-UV-ELSD串联色谱图。比对混合对照品色谱图中各色谱峰的保留时间,结果指认了峰3为槲皮素,峰4为芦丁,峰6为绞股蓝皂苷XLIX,峰8为绞股蓝皂苷A。最后以AIA格式将15批绞股蓝药材的HPLC-UV-ELSD指纹色谱图导入软件《中药色谱指纹图谱相似度评价系统(A版)》中,样品S1设为参照图谱,采用中位数对照图谱生成法,时间窗宽度设为0.2,指纹共有峰的叠加图谱分别如图1和图3所示,自动生成的对照指纹图谱分别见图2~4,参照峰为4号(芦丁)峰。最后确定了绞股蓝样品中有11个共有特征峰,其共有峰相对保留时间RSD分别为0.65%、0.68%、0.73%、0.00%、0.12%、0.55%、0.31%、0.76%、0.82%、0.67%、0.93%及相对峰面积RSD分别为10.23%、12.35%、13.63%、0.00%、11.53%、9.54%、10.27%、13.29%、15.82%、13.72%、10.70%。15批的绞股蓝样品与对照指纹图谱的相似度计算结果分别为0.999、0.990、0.991、0.989、0.986、0.995、0.992、0.993、0.991、0.995、0.996、0.988、0.987、0.990、0.990。
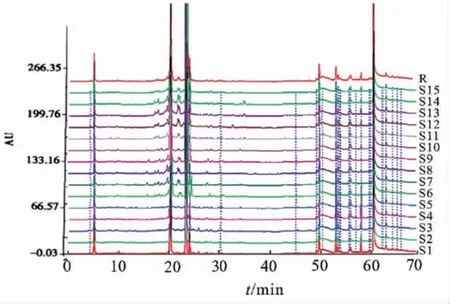
图1 15批绞股蓝药材HPLC-UV指纹图谱
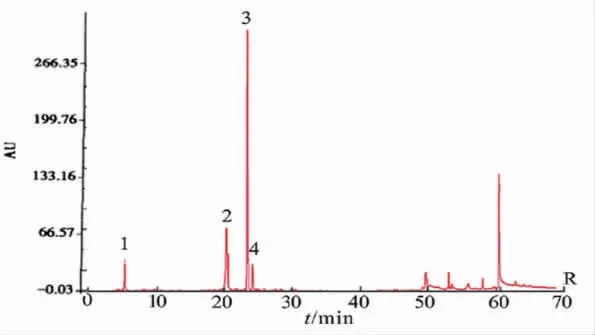
图2 15批绞股蓝药材HPLC-UV对照指纹图谱
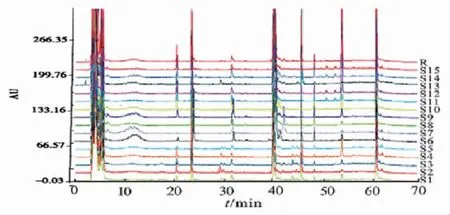
图3 15批绞股蓝药材HPLC-ELSD对照指纹图谱

图4 15批绞股蓝药材HPLC-ELSD对照指纹图谱
2.2 4 种成分的定量分析结果
2.2.1 线性关系考察 按已定的分析方法进样分析,绘制标准曲线,最后分别计算得线性回归方程为槲皮素Y1=4 502.7X1+38 912(r1=0.999 9);芦丁Y2=2 532.7X2+24 623(r2=0.999 8);绞股蓝皂苷AY3=6 477.7X3+47 010(r3=0.999 9);绞股蓝皂苷XLIXY4=6 482.7X4+47 720(r4=0.999 8)。结果表明,4种指标性成分在相应的线性范围内具有良好的线性关系。
2.2.2 精密度试验 取重复性试验中的同一供试品溶液连续进样6次。结果显示,含量RSD分别为槲皮素1.13%、芦丁1.25%、绞股蓝皂苷A 1.05%和绞股蓝皂苷XLIX 0.97%,表明本次试验所用的高效液相色谱仪的精密度良好。
2.2.3 稳定性试验 取同一供试品溶液,分别在0、2、4、8、12、24 h进样。结果显示,含量RSD分别为槲皮素0.33%、芦丁0.49%、绞股蓝皂苷A 0.72%和绞股蓝皂苷XLIX 0.35%,表明试验制备的供试品溶液24 h内稳定性良好。
2.2.4 重复性试验 分别精密称取6份过65目筛的绞股蓝药材粉末(S1)2.0 g,按照已定的样品制备方法进样分析检测。结果表明,槲皮素、芦丁、绞股蓝皂苷A和绞股蓝皂苷XLIXRSD分别为1.04%、0.55%、0.86%、1.13%,表明本次试验方法的重复性良好。
2.2.5 加样回收率试验 分别精密称取9份过65目筛的绞股蓝药材粉末(S1)样品0.1 g,将其平均分为3组(即50%、100%、150%),按已定样品制备方法进行制备,根据峰面积(A)计算平均加样回收率,结果显示,4种成分的平均加样回收率分别为槲皮素102.0%(RSD=1.27%)、芦丁98.5%(RSD=1.15%)、绞股蓝皂苷A 101.5%(RSD=0.89%)、绞股蓝皂苷XLIX 100.9%(RSD=0.99%)。
2.2.6 含量测定结果 取过65目筛的15批绞股蓝药材粉末,按已定方法制备和进样分析,分别计算药材中4种成分的含量,结果见表2。4种成分总含量高低顺序为:湖北十堰>陕西平利>陕西平利>湖北神农架>陕西龙西>云南昆明>安徽亳州>安徽亳州>云南昆明>福建古田>安徽亳州>广西桂林>安徽亳州>福建古田>安徽亳州。结果表明,湖北和陕西产地的绞股蓝药材含量均高于4.0%,可以归为Ⅰ类;云南、广西、福建、安徽产地的绞股蓝含量均低于4.0%,可以归为Ⅱ类。

表2 15批绞股蓝药材中4种成分的含量(%,n=3)
3 讨 论
3.1 提取方式考察 试验过程中还考察了不同提取方式对绞股蓝样品的提取效率,包括超声波提取、索氏加热回流提取、水浴加热回流提取,结果显示,索氏回流提取法所得色谱图的色谱峰信息较丰富,故试验选取索氏回流提取方法;查阅文献知[14-16],绞股蓝中可能含有大量的脂溶性成分,因此实验结合黄酮类成分(槲皮素、芦丁)和皂苷类成分(绞股蓝皂苷A、绞股蓝皂苷XLIX)的化学性质对提取溶剂,即石油醚(60~90℃)、三氯甲烷、甲醇、乙醇、50%甲醇、50%乙醇进行了考察,结果表明先以石油醚(60~90℃)除去干扰较大的脂溶性成分,再以甲醇为提取溶媒时色谱峰的丰度较好、基线较平稳、杂峰较少;对索氏回流提取时间,即提取1、2、4、6、8 h进行考察,结果显示索氏回流提取4 h即可提取完全。
3.2 色谱条件确定 本次实验通过全波长扫描(190~400 nm),最终选取最大吸光度360 nm作为本次实验的紫外检测波长;通过考察Agilent TC-C18、Thermo BDS C18和Agilent SB C18不同型号及不同厂家色谱柱,甲醇-0.1%乙酸水溶液、甲醇-0.5%乙酸水溶液、乙腈-0.5%乙酸水溶液、乙腈-0.1%乙酸水溶液、乙腈-0.1%甲酸水溶液、乙腈-水溶液、甲醇-0.1%甲酸水溶液和甲醇-水溶液不同流动相系统,25℃、30℃和40℃不同色谱柱柱温,0.8、1.0和1.2 mL/min不同流速,60℃、70℃和80℃不同漂移管温度。综合实验结果,最终选取Thermo BDS C18柱为本次实验的色谱柱、乙腈-0.5%乙酸水溶液为流动相、流速为1.0 mL/min、柱温为30℃、80℃的漂移管温度,在此条件下样品具有较丰富的色谱信息、平稳的基线、较高的丰度、较少的杂峰、平滑对称的峰形及较好的分离度。
3.3 小结 本次试验评价了15批不同产地绞股蓝药材的质量,结果建立了不同产地绞股蓝药材的HPLC-UV-ELSD指纹图谱,鉴定了共有特征峰11个,通过与对照品比对,指认了其中4个成分。在指纹图谱研究的基础上,同时定量分析了4个指标性成分的含量。由定量分析结果可知,不同产地绞股蓝药材中槲皮素、芦丁、绞股蓝皂苷A和绞股蓝皂苷XLIX的含量参差不齐,整体来看,不同产地绞股蓝样品中4种成分含量从高到低分别为:芦丁、绞股蓝皂苷A、绞股蓝皂苷XLIX和槲皮素。其中芦丁的平均含量约是槲皮素平均含量的10倍,差异较明显;黄酮类成分的总量是皂苷类成分的1.2倍,两大类成分总含量整体上相差不大。从产地上看,陕西和湖北产的绞股蓝药材质量较优,其他产地次之。本次试验建立的HPLC-UVELSD指纹图谱与多指标性成分定量分析方法简单、可靠,可作为绞股蓝药材质量的综合评价的方法之一,同时也为下一步建立完善的绞股蓝药材质量标准提供实验依据。
猜你喜欢
中成药(2018年1期)2018-02-02
中成药(2017年9期)2017-12-19
恋爱婚姻家庭·养生版(2017年12期)2017-12-07
恋爱婚姻家庭(2017年36期)2017-07-22
中成药(2017年3期)2017-05-17
陕西画报(2016年1期)2016-12-01
中国病理生理杂志(2015年8期)2015-12-21
药学研究(2015年11期)2015-12-19
医学研究杂志(2015年11期)2015-06-10
广西科技大学学报(2015年4期)2015-02-27
