
园林树木基因组学研究进展
2021-11-13 06:31林授锴林海兰陈璘霞温丽娟林顺权
莆田学院学报 2021年5期
林授锴,林海兰,陈璘霞,温丽娟,林顺权,3
(1.莆田学院 环境与生物工程学院,福建 莆田 351100;2.莆田市城市园林发展集团有限公司,福建 莆田 351100;3.华南农业大学 园艺学院,广东 广州 510642)
0 引言
园林树木是指适合在园林中栽培应用的木本植物。园林树木多数为具有观赏价值的树种,也包括不以美观见长但具有卫生防护和改善环境等作用的树种[1]。基因组学(genomics)是一门进行生物基因组研究和利用的学科,通过分析基因组DNA序列或其表达产物等来解读基因组信息,对于整合生命科学各学科分支,深化与开拓生命科学新的研究方向具有极其重大的意义[2]。
2000年拟南芥全基因组测序完成并公开,这是第一个基因组被完全测序的植物;2001年人类基因组计划公布了人类基因组草图;2002年水稻基因组测序完成[3]。园林树木的基因组学研究是在人类基因组计划和拟南芥、水稻等模式植物基因组计划取得成功的基础上开展起来的。
木本的模式植物毛果杨(Populus trichocarpa)是园林树木最早完成基因组测序的物种。2006年,Tuskan等测序并组装获得约485 Mb的毛果杨基因组序列,注释蛋白编码基因约45 000个;比对毛果杨(木本植物)和拟南芥(草本植物)基因组发现,有10%的毛果杨基因在拟南芥基因组中没找到对应部分[4]。园林树木在毛果杨测序完成之后有一个间断期,直到2012年我国学者完成了第一个木本花卉植物梅花(Prunus mune)的全基因组测序[5]。随后,学者们又陆续完成了马缨杜鹃(Rhododendron delavayi)[6]、樱花(Prunus yedoensis)[7]、月季(Rosa chinensis)[8]及其近缘种等的基因组测序。近年来,被测序的园林树木的种类越来越多。
本文将综述园林树木的基因组测序进展,介绍结构基因组学和功能基因组学的基本框架及其在园林树木上的应用进展,并对园林树木的基因组学研究进行展望。
1 园林树木基因组测序的主要进展
据笔者统计,截至2020年4月,至少已有175个科255种园艺植物被测序,表1选取其中的55种园林树木。
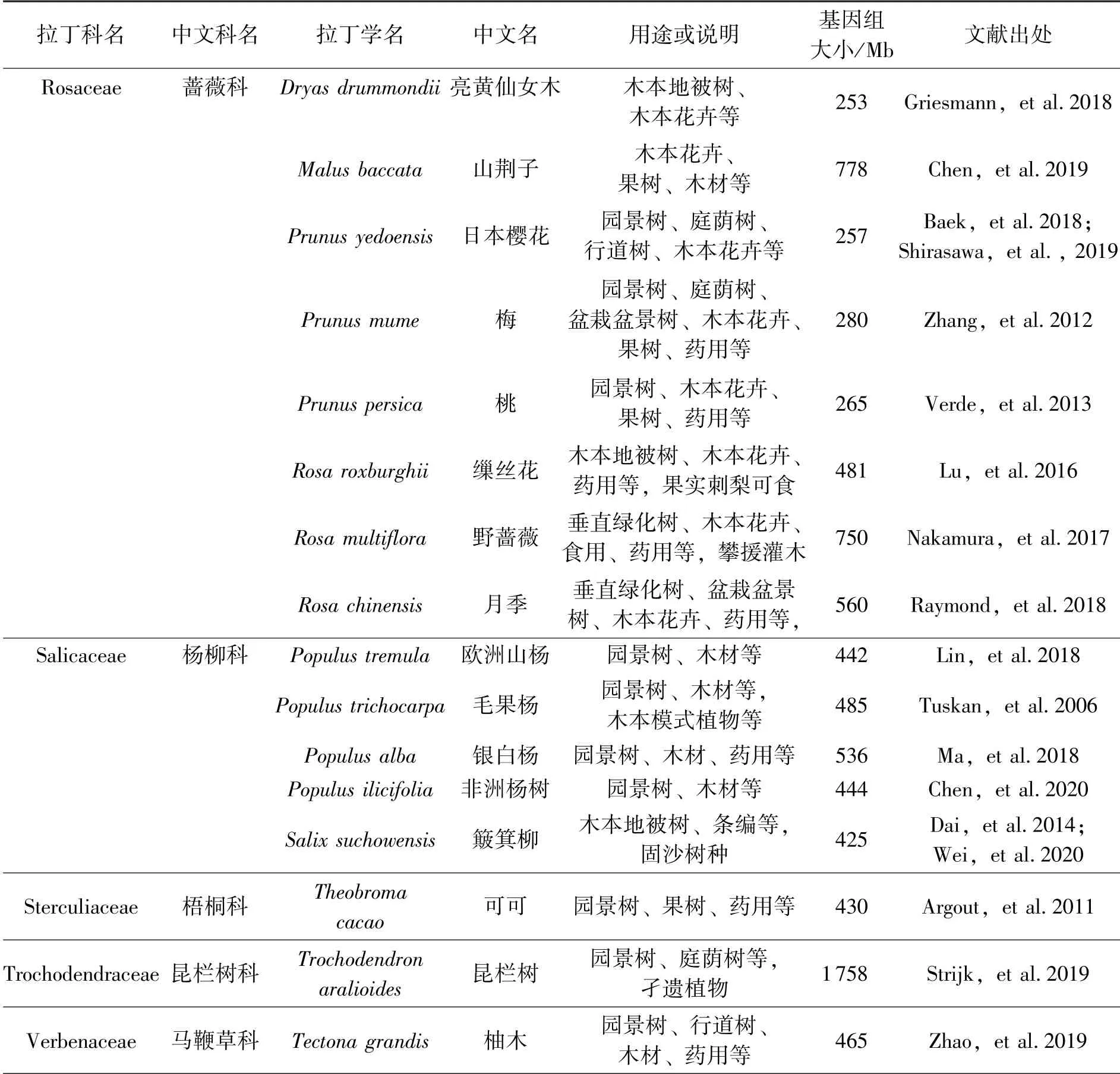
表1 (续)

表1 (续)
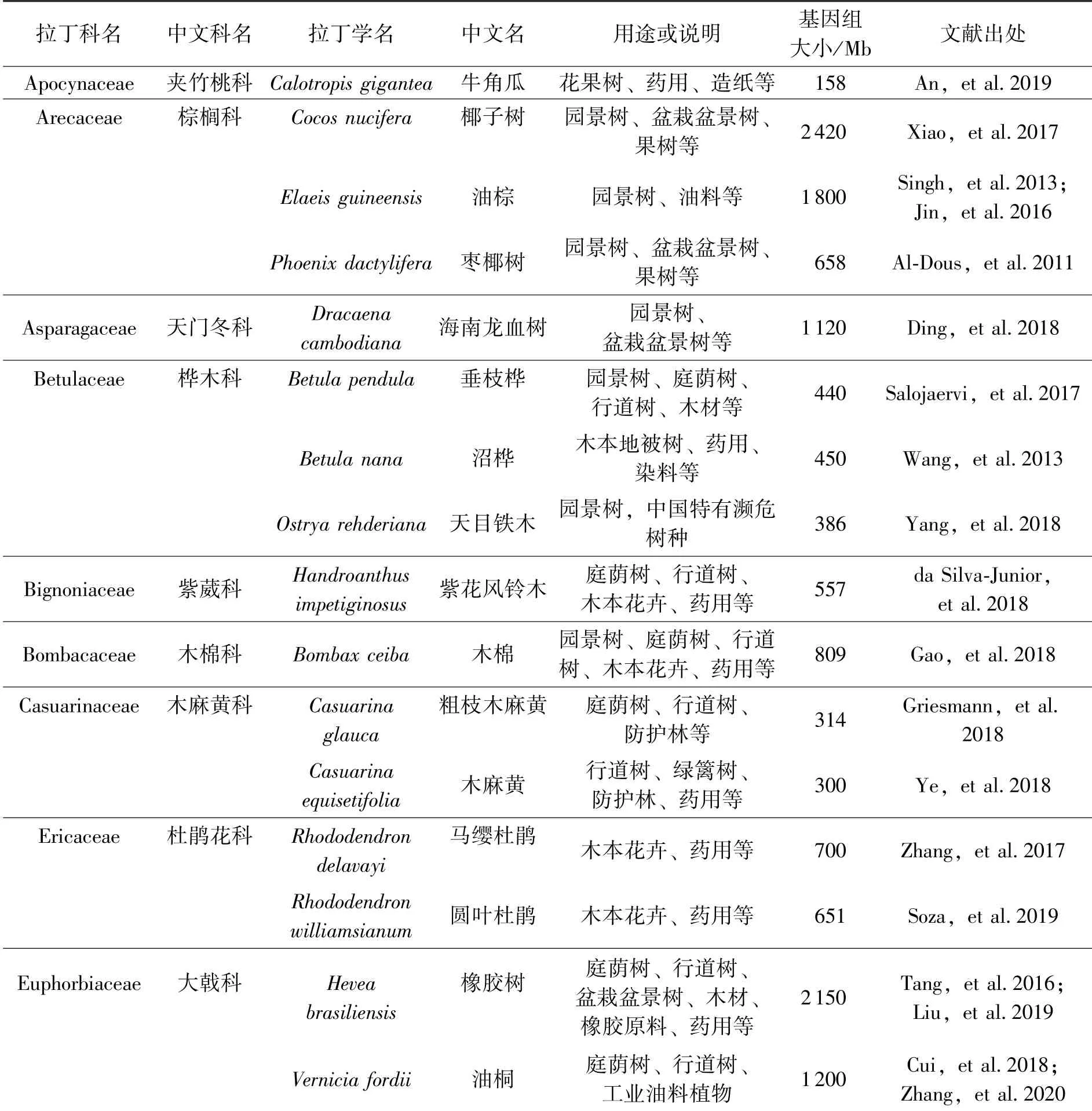
表1 全基因组序列已被发表的部分园林树木名录
2 园林树木结构基因组学与功能基因组学
园林树木基因组学研究可大致分为结构基因组学和功能基因组学两方面。
2.1 结构基因组学
结构基因组学以获得全基因组序列为目标,力求实现高效、准确的基因组测序。物种全基因组序列的获得是通过基因组测序与拼装来实现的,主要流程包括基因组的调查、测序、拼接、组装和组装质量评估[2]。
2.1.1 基因组调查
基因组调查,是在测序前对尚未进行基因组测序的物种的基因组状况进行调查,以评估目标基因组大小、倍性、杂合性等特征,为更好地选择合适的测序策略提供必要参考[2]。流式细胞术和高通量测序技术是最常用的两种基因组调查技术。段一凡等通过流式细胞术对14个桂花代表品种及其3个近缘种的倍性及基因组大小进行分析比对研究[9]。张月婷等通过高通量测序技术发现园林树种浙江楠基因组较大,且杂合率较高,需要利用二代与三代测序技术相结合的方式获得基因组[10]。陈双双等利用流式细胞术和高通量测序技术估测绣球主栽品种“Bailer”的基因组,发现绣球基因组较大且为杂合度较高的复杂基因组[11]。
2.1.2 基因组测序
Sanger法测序为第一代测序技术的主要代表,其对DNA片段的测序长度可达1000 bp,单个碱基的测序准确性高达99.99%,一直被广泛应用于生命科学领域。但其测序成本高、通量低、速度慢等缺点严重影响了其在基因组测序中的应用[12]。在园林树木中,仅毛果杨[4]、桃花[13]等极少数物种使用该法进行全基因组测序。
而自Sanger法测序诞生以来,经过近半个世纪的发展,测序技术取得了革命性进展。新产生的测序技术,根据出现时间、读长和通量等特征,被称作第二代、第三代测序技术等,也被统称为新一代测序技术[14]。
第二代测序技术又称高通量测序技术,以高通量、短读长和低成本为特征。绝大多数的高通量测序数据是由Illumina测序平台产生的,其测序准确性一般在99.5%以上[12]。通过纯二代测序获得基因组的园林树木有枣椰树[15]、梅花[5]等。但第二代测序技术存在GC偏好性及读长较短等缺陷,特别是对于具有高重复序列、高杂合、多倍体等特点的许多园林树木来说,在测序数据拼接环节中会受到一定的限制[2]。对此,早期研究者们采用了第二代结合第一代测序技术的策略加以应对,获得成功的园林树木有可可[16]等。
第三代测序技术是基于单个分子信号检测的DNA测序技术,完全跨过了第二代测序技术依赖于PCR扩增的信号放大过程,真正具备了读取单个荧光分子的能力。目前主流的第三代测序平台有PacBio公司的Sequel平台和Nanopore公司的PromethION平台等。第三代测序技术无碱基偏好性,读长可达8~25 kb,解决了第二代测序技术很多无法解决的问题,但是测序成本和测序错误率偏高。因此,在园林树木基因组测序中,第二代和第三代测序数据混合拼接的策略仍常被采用,以兼顾测序准确性、成本、拼接的连续性和拼接质量指标[12]。在园林树木中,垂枝桦[17]、小垫柳[18]等的基因组测序均采用第二代和第三代测序技术相结合的策略。近年来,随着第三代测序成本的不断下降及第三代测序数据的自纠错算法的不断发展,越来越多的园林树木仅采用第三代测序技术获取基因组序列,如月季[8]等。
2.1.3 基因组拼接
一个完整的基因组序列对目标物种的研究至关重要,但目前的基因组测序技术获得的序列普遍较短,到了现在的第三代测序技术也才8~25 kb,相对于一条染色体几十M(兆)的长度,如何通过短序列拼接获得高质量的全基因组序列是一个巨大的挑战。在目标物种全基因组序列排列顺序未知的情况下,需要通过计算机和特殊算法,把测序获得的序列拼接起来以获得基因组的真实序列,即从头拼接。基因组拼接的主要步骤包括:全基因组鸟枪法测序、原始序列质量控制、基因组从头拼接和基因组拼接质量提升[2,12]。限于篇幅,此处从略。
2.1.4 基因组组装
对于一个物种基因组,Scaffold水平的基因组草图是不够的,还需要通过添加额外的测序数据进行染色体水平的基因组组装。早期通常利用传统的遗传图谱来进行基因组的辅助组装,但使用遗传图谱辅助组装需要构建遗传群体,构图周期长且具有一定的难度。因此,许多新的技术(如Hi-C、Chicago、10×Genomics Linked-reads和BioNano光学图谱等)被开发出来应用于基因组的辅助组装[2,12]。多种辅助组装新技术也常常联用以获得尽可能高质量的染色体水平基因组,如使用BioNano光学图谱结合Hi-C技术将杜梨基因组组装到染色体水平[19],采用10×Genomics Linked-reads和Hi-C技术获得了蜡梅的染色体水平精细基因组[20]。
2.1.5 基因组组装质量评估
基因组组装完成后,需要对基因组质量进行评估,常见的方法有指标评估、一致性评估、完整性评估和准确性评估等。其中,指标评估主要评估Contig N50和Scaffold N50指标,指标越高说明基因组的连续性越好。准确性评估可以通过遗传图谱、BAC序列、Hi-C和光学图谱等其他类型的数据与组装结果进行比对来验证组装结果的正确性,其一致性越高,基因组越准确[2,12]。
2.2 功能基因组学
功能基因组学利用结构基因组学提供的序列信息,在基因组水平上进行高通量的序列分析和基因功能鉴定。功能基因组学的研究内容主要包括基因组注释、功能基因定位、候选功能基因分析和基因功能验证等[2,12]。
2.2.1 基因组注释
在基因组测序组装完成后,首先就要对基因组进行注释。基因组注释是利用生物信息学方法和工具,对基因组中所有基因的结构进行鉴定(基因组结构注释),并对基因的生物学功能进行注释(基因组功能注释)。一个完整的注释包括在基因组中鉴定出其功能元件(蛋白质编码基因、RNA基因、重复序列和假基因等),并尽可能地确定这些元件所对应的生物学功能[21]。限于篇幅,此处从略。
2.2.2 功能基因定位
目前在植物中常用的功能基因定位方法有两种:一种是从上而下的方法,通过表型差异来找到对应的突变基因并揭示其功能,包括连锁分析、全基因组关联分析(genome-wide association study,GWAS)、混池分离分析(bulked segregant analysis,BSA)等;另一种是从下而上的方法,通过检测基因组上的选择信号来定位功能基因,包括基因组选择信号分析等[12]。
连锁分析是利用连锁的原理研究基因与遗传标记的关系。基于连锁分析,基因或者遗传标记在染色体上以一定的顺序排列构成一张遗传图谱。遗传图谱是进行植物基因组结构分析与功能研究的基础[12]。一些园林树木已构建了高质量的遗传图谱,如梅花[22]、桂花[23]和月季[24]等。
GWAS主要在全基因组水平分析各位点与复杂性状遗传变异的关联强弱。它以连锁不平衡为基础,通过分析数百或数千个体的高密度SNP分子标记的分离特征,筛选出与复杂性状表现型变异相关联的分子标记,进而分析这些分子标记对表现型变异的遗传效应[2,12]。文[25]应用GWAS定位了园林树木毛果杨与发芽时间有关的关键位点;文[26]应用GWAS定位了大叶桉树高性状的SNP位点;文[27]利用GWAS进一步确定了桂花橙色花性状相关的SNP位点和候选基因。
BSA是利用极端表型个体进行功能基因挖掘的一种定位方法。随着第二代测序技术的发展,SNP标记开发和测序成本不断下降,BSA技术已成为连锁分析和GWAS以外的另一个定位功能基因的有效工具。不同于连锁分析和GWAS,BSA基于表型分组,可以分别针对质量性状主效基因和数量性状基因座(quantitative trait locus,QTL)进行定位。对主效基因定位,可以简单基于两个差异表型进行分组;对于QTL定位,可以基于极端表型群体混合并分组[2,12]。BSA技术已被应用于北方黑云杉[28]等少数园林树木的QTL定位。
在进化过程中,自然或人工选择在基因组上留下了各种选择印迹,这些印迹称为选择信号。选择主要包括正选择、负选择和平衡选择等3个类型。其中正选择是生物适应性进化的主要机制,且往往在基因组上留下明显的选择信号[12]。文[29]便应用基因组选择分析,揭示了毛果杨在广泛纬度范围内适应性变异的基因组基础。
2.2.3 候选功能基因分析
在基因组水平上有一系列方法可以定位功能基因,但均具有一定的局限性,因此需要综合其他数据进一步分析以缩小候选功能基因范围。随着高通量测序技术、质谱技术和表型测量技术的发展,大规模获取转录组、蛋白质组、代谢组和表型组数据都为候选功能基因分析提供了强有力的支撑[12]。如综合基因组和转录组数据,挖掘到了控制梅花花瓣颜色的候选功能基因MYB108[30]。
2.2.4 基因功能验证
基因功能研究大致可以分为正向遗传学和反向遗传学两类。正向遗传学是从表型变化研究基因变化,反向遗传学则是从基因变化研究表型变化。反向遗传学实验技术通过对特定基因的敲除、敲入、过表达或沉默来寻找关联表型,进而分析基因的功能。对于通过功能基因定位、候选功能基因分析等技术寻找并筛选出的候选功能基因,往往会采用基因编辑、过表达或沉默等反向遗传学基因功能分析技术来对其基因功能进行验证[12]。
然而,能进行稳定遗传转化获得转基因植株的园林树木仍然较少,并且实验周期较长。如一项为期近20年的研究工作发现,在3种杨树中过表达毛果杨SVL基因可以显著延迟杨树开花,并减少花芽丰度[31]。对于许多遗传转化体系还不成熟的园林树木,验证基因功能往往在拟南芥等模式植物中进行异源转化。如在拟南芥中过表达胡杨PeuLAC2基因可以增强植物抗氧化酶活性、降低活性氧含量、提高茎水势、提高植物对水分的运输能力,从而增强植物的抗旱能力[32]。
3 展望
毛果杨作为木本模式植物2006年便被测序,成为了最先被测序的园林树木。数年后才有人完成其他一些园林树木的全基因组测序,而且主要是具有观赏价值的果树,如枣椰树、梅树、桃树等。可喜的是,我国科学家率先发表梅花等的全基因组序列。近年来随着测序技术的迅猛发展和测序成本的大幅降低,研究者对越来越多的园林树木进行了全基因组测序。
在结构基因组学研究上,由于许多园林树木测序时间较晚,基于模式植物、果树等的测序经验,以及使用更为先进的测序与组装技术,多数园林树木的全基因组测序结果较为理想。随着越来越多的园林树木获得研究者的关注,对缺乏参考基因组的园林树木进行测序以获得全基因组序列,是园林树木基因组学研究的一个重要方向。
结合结构基因组研究,一些基本的功能基因组研究往往同时开展,如基因组功能注释、基于自然群体的基因定位和少数候选功能基因的挖掘。除此以外,多数园林树木功能基因组学的研究进展是缓慢的,主要原因在于:一是园林树木不但基因高度杂合,而且有性生殖周期很长,遗传群体构建困难,这与果树类似;二是除模式植物毛果杨等少数树种外,多数园林树木受到的关注较少,再生体系尚未建立,使得其功能验证受限。因此,对园林树木功能基因组学的研究,要充分开发和利用复杂遗传群体非依赖性的基因定位和挖掘技术,并在不同园林树木树种的再生体系上争取突破。
总体而言,除了木本模式植物毛果杨外,园林树木的基因组学研究进展整体上是慢于果树的,这或许与人们更加关注果树的经济效益有关。植物基因组学研究的先进水平,与物种的重要性息息相关。模式植物拟南芥是最先进的,然后是粮食作物,之后才是包括园艺植物在内的其他经济作物,过去是这样,将来仍将是这样。但是,在未来,园艺植物内部可能会发生变化,过去是果树较为先进,但是随着城市化进程的加快,加上一些园林树木在离体再生方面比果树更方便,整体上园林树木的基因组学和分子生物学研究进展有可能追上甚至超过果树。
园林树木不但具有丰富多彩的观赏性状,特别是开花性状方面具有高度的多样性,而且其生态价值在未来必将得到更大的重视,因此,有理由相信将会有更多的研究者致力于园林树木的基因组研究。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
军事文摘(2022年16期)2022-08-24
今日农业(2022年4期)2022-06-01
中国典型病例大全(2022年7期)2022-04-22
今日农业(2021年14期)2021-10-14
今日农业(2021年11期)2021-08-13
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国中药杂志(2016年21期)2017-02-16
中国中药杂志(2016年21期)2017-02-16
