
本草基因组学
2017-02-16 13:20陈士林宋经元
中国中药杂志 2016年21期
陈士林+宋经元

[摘要]中醫药学对世界医药学发展作出了巨大贡献,随着现代科学技术的发展,特别是人类基因组计划的提出和完成,对人类疾病的认识和治疗开启了全新篇章。在此背景下,笔者将组学技术引入中药学研究,提出本草基因组学(Herbgenomics)学科概念,即利用组学技术研究中药基原物种的生物遗传信息及其调控网络,阐明中药防治人类疾病分子机制的学科,从基因组水平研究中药及其对人体作用的前沿科学。主要内容涉及结构基因组、功能基因组、蛋白质组、转录组、代谢组、表观基因组、宏基因组、药用模式生物、基因组辅助分子育种、DNA鉴定、中药合成生物学、中药基因组学、生物信息学及数据库等理论与实验技术。本草基因组学为中药药性研究提供理论基础,为中草药次生代谢产物的生物合成和代谢工程提供技术支撑,为中药配伍研究提供科学依据,指导药物开发及合理用药,为实现个体化精准医疗提供重要信息和技术保障,为中药道地品种改良和基因资源保护奠定基础,推动中药农业的科学发展,对培养多学科人才充实到传统药物研究具有引领作用。本草基因组学正促进前沿生命科学技术应用到中药领域,对中药现代化进程具有重大战略性科学意义。
[关键词]本草基因组学; 基因组学; 组学; 中药
[Abstract]Traditional Chinese medicine (TCM) has contributad greatly to improving human health However, the biological characteristics and molecular mechanisms of TCM in the treatment of human diseases remain largely unknown Genomics plays an important role in modern medicine and biology Here, we introduce genomics and other related omics to the study of herbs to propose a new discipline, Herbgenomics, that aims to uncover the genetic information and regulatory networks of herbs and to clarify their molecular mechanisms in the prevention and treatment of human diseases Herbgenomics includes herbal structural genomics, functional genomics, transcriptomics, proteomics, metabonomics, epigenomics and metagenomics Genomic information, together with transcriptomic, proteomic, and metabolomic data, can therefore be used to predict secondary metabolite biosynthetic pathways and their regulation, triggering a revolution in discoverybased research aimed at understanding the genetics and biology of herbs Herbgenomics provides an effective platform to support chemical and biological analyses of complex herbal products that may contain more than one active component Herbgenomics is now being applied to many areas of herb related biological research to help understand the quality of traditional medicines and for molecular herb identification through the establishment of an herbal gene bank Moreover, functional genomics can contribute to model herb research platforms, geoherbal research, genomicsassisted herb breeding, and herbal synthetic biology, all of which are important for securing the future of medicinal plants and their active compounds In addition, Herbgenomics will facilitate the elucidation of the targets and mechanism of herbs in disease treatment and provide support for personalized precise medicineHerbgenomics will accelerate the application of cuttingedge technologies in herbal research and provide an unprecedented opportunity to revolutionize the use and acceptance of traditional herbal medicines
[Key words]Herbgenomics; genomics; omics; traditional Chinese medicine (TCM)
doi:10.4268/cjcmm20162101
本草基因组学(herbgenomics)是利用组学技术研究中药基原物种的遗传信息及其调控网络,阐明中药防治人类疾病分子机制的学科,从基因组水平研究中药及其对人体作用的前沿科学。涉及中草药结构基因组、中草药转录组、中草药功能基因组、中草药蛋白质组、中药代谢组、中草药表观基因组、中草药宏基因组、药用模式生物、基因组辅助分子育种、DNA鉴定、中药合成生物学、中药基因组学、中草药生物信息学及数据库等理论与实验技术。
传统药物应用历史悠久,应用方式多样,相关研究主要集中在形态识别、化学物质基础揭示、药效作用分析、资源调查、人工栽培等方面,但长期以来对传统药物基因资源的认识和了解十分薄弱,人才极其匮乏。由于中药原植物基因组信息缺乏,中医药学和现代生命科学之间缺乏沟通的桥梁,新兴的前沿生命科学技术很难应用于传统中医药研究,如对于中药道地性形成和维持的遗传机制及道地性和药性的相互关系缺乏深入了解,已严重影响了我国道地药材的资源保护和新品种选育,中药道地性形成和维持的遗传基础研究急需加强;中药药性的生物学本质研究亟待加强,多年来中药药性研究主要集中在化学和药理方向,但对于中药药性的生物学本质研究还非常薄弱,已从根本上制约了对中药药性的深入研究;中药基因资源是一种珍贵的国家战略资源,国际竞争严峻,韩国、美国、日本等国家已启动许多中药基原物种全基因组研究,对我国传统中药研究领域造成极大挑战。另外,由于大多数药用植物有效成分含量低,分离提取需要消耗大量原料,对天然资源造成极大破坏,也使得多数提取类药物的生产成本很高。
本草基因组学作为新兴学科,广义而言是从基因组水平研究中药及其对人体作用。一方面从基因组水平研究基因序列的多态性与药物效应多样性之间的关系,研究基因及其突变体对不同个体药物作用效应差异的影响,从蛋白质组学角度研究中药作用靶点,特别是中药复方的多靶点效应,为中药配伍提供科学依据,指导药物开发及合理用药,为实现个体化精准医疗提供重要信息和技术保障;另一方面建立含有重要活性成分的中药原植物基因组研究体系,系统发掘中药活性成分合成及优良农艺性状相关基因,解析代谢物的合成途径、代谢物网络及调控机理,为中药道地品种改良和基因资源保护奠定基础,为中药药性研究提供理论基础,对传统药物学理论研究和应用具有重要意义,从基因组层面阐释中药道地性的分子基础,推动中药创新药物研发,为次生代谢产物的生物合成和代谢工程提供技术支撑,创新天然药物研发方式,为优质高产药用植物品种选育奠定坚实基础,推动中药农业的科学发展,对揭示天然药物形成的生物学本质具有重要价值,对培养多学科人才充实到传统药物研究具有引领作用。狭义而言本草基因组学集中研究中草药本身的遗传信息,不涉及对人体的作用。也就是说狭义本草基因组学主要研究中草药结构基因组、转录组、功能基因组、蛋白质组、代谢组、表观基因组、宏基因组,以揭示中药道地性和中药药性的遗传本质。本草基因组学正促进前沿生命科学技术应用到中药领域,推动中药研究迅速走到生命科学的最前沿。
1 本草基因组学的产生和发展
1.1 本草基因组学的产生 从“神农尝百草,一日而遇七十毒”的传说到现存最早的中药学著作《神农本草经》(又称《本草经》),从世界上现存最早的国家药典《新修本草》(即《唐本草》)到本草学巨著《本草纲目》,两千多年来,中药学的发展反映了我国劳动人民在寻找天然药物、利用天然药物方面积累了丰富经验。中药学是中国医药学的伟大宝库,对世界医药学发展作出了巨大贡献。随着现代科学技术的发展,特别是人类基因组计划(Human Genome Project)的提出和完成,对人类疾病的认识和治疗开启了全新的篇章,在此背景下,中药学研究逐渐深入到基因组水平从而导致本草基因组学产生和兴起。
1977年Sanger完成首个物种全基因组测序,噬菌体φX174基因组,大小为5.836 kb[1];人类基因组计划由美国科学家于1985年率先提出,1990年正式启动,2000年完成,是一项规模宏大,跨国跨学科的科学探索工程,其宗旨在于测定组成人类染色体(指单倍体)中所包含的30亿个碱基对组成的核苷酸序列,从而绘制人类基因组图谱,并且辨识其载有的基因及其序列,达到破译人类遗传信息的最终目的[2-3]。2000年,破译拟南芥Arabidopsis thaliana全基因组,大小为125 Mb,作为第一个植物全基因组测序在植物科学史上具有里程碑意义[4]。我国药用植物有11 146种,约占中药材资源总数的87%[5],是所有经济植物中最多的一类。同时,药用植物也是許多化学药物的重要原料,目前1/3以上的临床用药来源于植物提取物或其衍生物,其中最著名的青蒿素来源植物是黄花蒿。
中国学者应用光学图谱和新一代测序技术,完成染色体水平的灵芝基因组精细图绘制,通过基因组解析提出灵芝为首个中药基原的药用模式真菌,文章发表在《自然通讯》上,期刊编辑部以特别图片(featured image)形式进行了推介(图1)[6],认为该论文表明灵芝对于研究传统菌类中药的次生代谢途径及其调控是一个有价值的模式系统。灵芝基因组图谱的公布为开展灵芝三萜等有效成分的合成研究提供了便利,随着这些合成途径的逐步解析,使得通过合成生物学合成灵芝有效成分成为可能。同时,对灵芝生长发育和抗病抗逆关键基因的发掘和认知,将推动灵芝的基因组辅助育种研究,加速灵芝新品种的培育,并为灵芝的科学栽培和采收提供理论指导。
2009年,陈士林团队提出本草基因组计划,即针对具有重大经济价值和典型次生代谢途径的药用植物进行的全基因组测序和后基因组学研究,全基因组测序、组装和分析策略:测序物种的筛选原则,待测物种基因组预分析,测序平台的选择,遗传图谱和物理图谱的绘制,全基因组的组装及生物信息学分析;模式药用植物突变体库的建立和基因功能研究;药用植物有效成分的合成及其调控研究;药用植物抗病抗逆等优良性状的遗传机制研究及优良品种选育。在此基础上,详细介绍了本草基因组方法学研究:全面介绍物种基因组大小、染色体数目测定方法、第二代高通量测序方法、全基因组组装和基因组注释方法、基因组比较等生物信息学分析手段、简要阐述重测序在药用植物全基因组研究中的应用方法。由此,本草基因组学逐渐形成和完善,包括中草药结构基因组、转录组、功能基因组、蛋白质组学、代谢组、表观基因组、宏基因组、基因组辅助分子育种、中药合成生物学、中药基因组学、中草药生物信息学及数据库等内容。基于分子生物学和基因组学的药用植物鉴别是当前研究的活跃领域,用于鉴别的分子生物学和基因组学技术:AFLP、RFLP、RAPD、DNA微阵列技术(microarray)、DNA条形码(barcoding)等,基于基因组鉴别的分子基础是植物分子系统发育关系反映物种进化关系。在这些技术当中,药用植物DNA条形码鉴定策略及关键技术是最受关注的方向,中药材DNA条形码分子鉴定指导原则已列入《中国药典》2010年版增补本Ⅲ和《中国药典》2015年版。
1.2 本草基因组学的发展 2015年国际期刊《科学》增刊详述“本草基因组解读传统药物的生物学机制”,提出本草基因组学为药用模式生物、道地药材研究、基因组辅助育种、中药合成生物学、DNA鉴定、基因数据库构建等提供理论基础和技术支撑(图2)。目前,药用植物基因组学与生物信息学已经进入快速发展阶段,必将对传统药物学产生巨大影响。国内外已经开展青蒿[7]、丹参[8-15]、西洋参[16]、甘草[17]等多种药用植物的大规模转录组研究。基因组序列包含生物的起源、进化、发育、生理以及与遗传性状有关的一切信息,是从分子水平上全面解析各种生命现象的前提和基础。第二代高通量测序技术的飞速发展及第三代单分子测序技术的兴起使测序成本大大降低,测序时间大大缩短,为本草基因组计划的实施奠定了坚实的技术基础。目前,赤芝[6]、紫芝[18]、丹参[19]及铁皮石斛[20-21]等重要药用植物的基因组已完成测序工作并发表,人参、苦荞、穿心莲、紫苏等中草药基因组图谱也完成绘制。
例如为了解析丹参的遗传背景,陈士林团队联合国内外著名高校和研究机构,通过联合测序技术完成了丹参基因组图谱的组装,丹参基因组的完成代表着首个鼠尾草属物种基因组图谱的成功绘制。进化分析显示丹参与芝麻亲缘关系更近,估计其分化时间约6 700万年前。丹参基因组的发表推动首个药用模式植物研究体系的确立。本草基因组学将开辟中药研究和应用的全新领域,把握历史性机遇,将极大提高我国开发中药资源的能力,增强我国中药基础研究实力、提高我国中药研究的自主创新能力,对于加速中药现代化进程具有重大的战略性科学意义,促进中药研究和产业的快速发展[22]。本草基因组学将使中草药生物学研究进入一个崭新的时代——本草基因组时代。
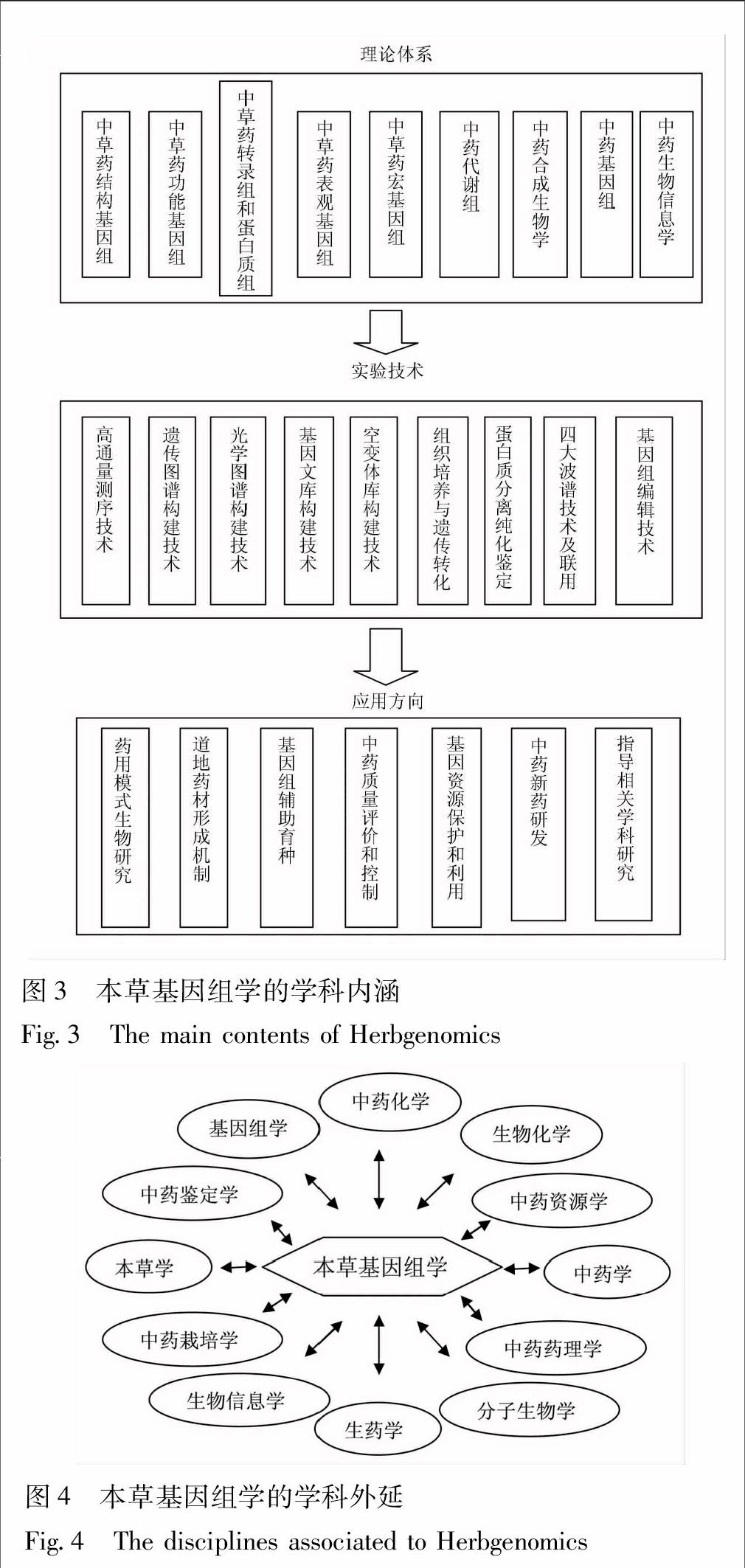
1.3 学科内涵和外延 根据本草基因组学产生和发展过程,主要从3个方面确定学科的内涵,即理论体系、实验技术和应用方向(图3)。本草基因组学形成了高度综合的理论体系,包括从基因组水平研究本草的九大内容:中草药结构基因组、中草药功能基因组、中草药转录组和蛋白质组、中药代谢组、中草药表观基因组、中草药宏基因组、中药合成生物学、中药基因组学、中草药生物信息学等。本草基因组学的实验方法主要包括九大技术:高通量测序技术、遗传图谱构建技术、光学图谱构建技术、基因文库构建技术、突变库构建技术、组织培养与遗传转化、蛋白质分离纯化与鉴定技术、四大波谱技术及联用、基因组编辑技术等。基于本草基因组学的理论体系和实验技术,形成了该学科的七大应用方向:药用模式生物研究、阐明道地药材形成机制、基因组辅助育种、基因资源保护和利用、中药质量评价和控制、中药新药研发、指导相关学科研究。
本草基因组学的学科外延与本草学、中药学、基因组学、生物信息学、分子生物学、生物化学、生药学、中药资源学、中药鉴定学、中药栽培学、中药药理学、中药化学等密切相关(图4)。本草学和中药学为本草基因组学奠定了深厚的历史基础和人文基础,为本草基因组学研究对象的确定提供丰富候选材料,基因组学和生物信息学为本草基因组学提供前沿理论和技术支撑,分子生物学、生物化学、中药化学则为本草基因组学提供基础理论和基本实验技术支持,生药学、中药资源学、中药鉴定学、中药栽培学与本草基因组学互相支撑发展,各学科的侧重点不同,中药药理学、中药化学为本草基因组学的应用提供技术支持。与以上各学科相呼应,本草基因组学促进本草学和中药学从经典走向现代、从传统走向前沿,为中医药更好服务大众健康提供强大知识和技术支撑,扩大了基因组学和生物信息学的研究对象和应用领域,为分子生物学、生物化学、中药化学走向实践应用提供了生动案例,推动生药学、中药资源学、中药鉴定学、中药栽培学从基因组和分子水平开展研究,为中药药理学的深入研究提供理论和技术支持。
2 本草基因组学研究內容
本草基因组学借助基因组学研究最新成果,开展中草药结构基因组、中草药功能基因组、中草药转录组和蛋白质组、中草药表观基因组、中草药宏基因组、中药合成生物学、中药代谢组、中药基因组学、中草药生物信息学及数据库等理论研究,同时对基因组研究相关实验技术在本草学中的应用与开发进行评价,推动本草生物学本质的揭示,促进遗传资源、化学质量、药物疗效相互关系的认识,以下详细阐述本草基因组学的研究内容。
2.1 中草药结构基因组研究 我国药用资源种类繁多,因此药用物种全基因组计划测序物种的选择应该综合考虑物种的经济价值和科学意义,并按照基因组从小到大、从简单到复杂的顺序进行测序研究。在测序平台的选择上应以第二代及第三代高通量测序平台为主,以第一代测序技术为辅。近年来,紫芝、赤芝、茯苓、丹参、人参、三七等10余种药用植物被筛选作为本草基因组计划的第一批测序物种,其中赤芝结构基因组发表被《今日美国》(USA Today)以“揭秘中国‘仙草基因组”为题报道(图5),丹参基因组小(约600 Mb)、生长周期短、组织培养和遗传转化体系成熟等原因,被认为是研究中药活性成分生物合成理想的模式植物[23]。丹参全基因组测序完成已推动丹参作为第一个药用模式植物研究体系形成。
由于多数药用植物都缺乏系统的分子遗传学研究,因此在开展全基因组计划之前进行基因组预分析非常必要。基因组预分析的主要内容包括:①利用条形码等技术对满足筛选原则的待测物种进行鉴定[24-25];②通过观察有丝分裂中期染色体确定待测物种的染色体倍性和条数;③采用流式细胞术[26]或脉冲场电泳技术估测物种的基因组大小,为测序平台的选择提供参考;④基因组Survey测序,在大规模全基因组深度测序之前,首先对所选药用植物进行低覆盖度的Survey测序,用来评价其基因组大小、复杂度、重复序列、GC含量等信息。
遗传图谱和物理图谱在植物复杂的大基因组组装中具有重要作用。借助于遗传图谱或物理图谱中的分子标记,可将测序拼接产生的scaffolds按顺序定位到染色體上。但遗传图谱的构建需要遗传关系明确的亲本和子代株系,因此其在大多数药用植物中的应用受到限制。物理图谱描绘DNA上可以识别的标记位置和相互之间的距离(碱基数目)。最初的物理图谱绘制多是基于BAC文库,通过限制性酶切指纹图谱、荧光原位杂交等技术将BAC克隆按其在染色体上的顺序排列,不间断地覆盖到染色体上的一段区域[27]。如今,光学图谱OpGen[28]和单分子光学图谱BioNano等[29]依赖于大分子DNA酶切标记的方法常用于物理图谱的绘制。
随着第二代测序技术的快速发展,用于短序列拼接的生物信息学软件大量涌现,常用软件包括Velvet[30], Euler[31], SOAPdenovo2[32], CAP3[33]等。基因组草图组装完成后,可利用生物信息学方法对基因组进行分析和注释,为后续功能基因组研究提供丰富的资源。例如,可以通过GeneScan[34], FgeneSH[35]等工具发现和预测基因,利用BLAST同源序列比对或InterProScan[36]结构域搜索等方法对基因进行注释,利用GO分析对基因进行功能分类[37],利用KEGG对代谢途径进行分析等[38]。
2.2 中草药功能基因组研究 根据全基因组序列和结构信息,中草药功能基因组研究充分利用转录组学、蛋白组学、代谢组学等方法,对药用植物的功能基因进行发掘和鉴定,研究内容主要集中于构建模式药用植物平台、次生代谢产物合成途径和调控机制的解析、抗病抗逆等优良农艺性状遗传机制的揭示等。
拟南芥、水稻等重要模式植物均具有大规模的T-DNA 插入突变体库,利用这些突变体库发掘了大量生长发育、抗逆性、代谢相关的重要基因。丹参等模式药用植物全基因组序列和大规模突变体库的建立将为药用植物研究提供丰富的资源和材料,从而推动药用植物功能基因研究, 尤其是次生代谢途径相关基因的鉴定进程,突变体库中的一些具有抗逆、抗病、高产等优良性状的突变株系以及转基因植株也是良好的新种质资源。药用植物有效成分的生物合成途径和调控方面的研究还很薄弱,主要集中在长春花、青蒿和甘草等少数物种,一些具有重大商业价值的天然药物,如紫杉醇、长春碱、喜树碱等生物合成途径至今还未被完全解析,已有报道多采用单基因研究策略。本草基因组学为次生代谢途径相关基因的“批量化”发掘奠定基础,对次生代谢产物的生物合成及代谢工程等应用领域产生重要影响。
与生长发育、抗逆抗病、重要遗传性状及种质性状控制相关的基因是药用植物重要的功能基因,利用基因组注释信息,发掘优良基因,运用基因工程的手段打破生殖隔离,培育活性成分含量高的具有优良农艺性状的新品种,为活性成分的大量提取和广泛临床应用奠定基础[39]。中草药结构基因组将为转录组分析和基因组重测序研究提供参考序列,通过对种内或品种间种群个体的转录组测序和重测序可快速、准确、大规模地发现SNP,SSR,InDel等分子标记,加速分子标记和优良性状的遗传连锁研究,快速发现药用植物的表型、生理特征与基因型的关系,提高育种工作效率[39]。
2.3 中药组学其他研究 中草药转录组学是中草药功能基因组学的重要研究内容,是在整体水平上研究中草药某一生长阶段特定组织或细胞中全部转录本的种类、结构和功能以及基因转录调控规律的科学。中草药转录组研究为鉴定中草药植物生长发育及抗病抗逆等优良性状相关的基因功能提供基础[40-41]。目前,在多数中草药植物无法进行全基因组测序的情况下,转录表达谱研究成为比较基因序列、鉴定基因表达的一种快速方法。通过对中草药不同组织部位、不同生长时期、不同生长环境下的转录组进行比较分析,可有效发掘参与中草药植物生长发育及抗病抗逆等优良性状相关基因。
中药蛋白质组学是将蛋白质组学技术应用于中药研究领域,一方面通过比较对照细胞或动物组织的蛋白质表达谱和给予中药后蛋白质表达谱的差异,可找到中药的可能靶点相关蛋白质,另一方面不同中草药及其不同组分例如根茎叶中蛋白质组的差异,以评价中草药活性成分与其生长过程中蛋白组变化的关系,寻找中药高活性的机制。不同于其他蛋白质组学,中药蛋白质组学的研究对象为中草药本身及用中药(单体化合物、中药组份或复方)处理后的生物体(细胞或组织),发现中药的有效成分及作用机制。中药蛋白质组学的研究目标包括:中药药物作用靶点的发现和确认,特别是中药复方的多靶点效应,蛋白质组学能更好发现中药复方的多种靶点,研究中药植物蛋白质组成差异,阐明中药作用机制及中药毒理作用机制,以及为中药配伍提供科学依据。
中药代谢组学结合中草药结构基因组解析代谢物的合成途径、代谢物网络及调控机理,研究内容主要包括药用植物的鉴别和质量评价,药用植物品种选育及抗逆研究,初生、次生代谢途径解析,代谢网络、代谢工程研究及合成生物学研究等几个方面,最终为药用植物品种选育、创新药物研发和质量安全性评价奠定基础。
中药基因组学从基因水平研究基因序列的多态性与药物效应多样性之间的关系,研究基因及其突变体对不同个体药物作用效应差异的影响,以此平台指导药物开发及合理用药,为提高药物的安全性和有效性,避免不良反应,减少药物治疗费用和风险,实现个体化精准医疗提供重要信息和技术保障。例如,Sertel等[42]经基因检测得出53/56的基因上游位置包含一个或多个c-Myc/Max结合位点,c-Myc和Max介导的转录控制基因表达可能有助于提高青蒿琥酯对癌细胞的治疗效果[43]。又如,银杏具有显著的诱导CYP2C19活性效应,研究显示不同CYP2C19基因型个体,银杏与奥美拉唑(omeprazole,广泛使用的CYP2C19底物)存在潜在的中西药互作关系。Chen等 [44]研究了健康志愿者体内六味地黄丸潜在的中-西药相互作用以及是否受基因型影响。
中草药表观基因学是针对本草基因组计划中具有重要经济价值的药用植物和代表不同次生代谢途径的模式药用植物开展表观基因组学研究。研究内容主要包含4个领域:分别是DNA甲基化、蛋白质共价修飾、染色质重塑、非编码RNA调控。中草药表观基因组学将通过研究重要中药材(药用生物)的基因组信息及其表观遗传信息变化,探索环境与基因、基因与基因的相互作用,解析哪些基因受到环境因素的影响而出现表观遗传变化可能提高中药材的药效品质,哪些表观遗传信息影响中药的性味等。
中草药宏基因组学是以多种微生物基因组为研究对象,对药材生长环境中微生物的多样性、种群结构、进化关系、功能活性以及微生物与药材生长相互协作关系进行研究的一门学科,对于帮助解决中草药连作障碍等现实问题具有重要指导作用。
药用模式生物研究体系的确立是本草基因组学的重大贡献,该体系具有模式生物的共同特征。从一般生物学属性上看,通常具有世代周期较短、子代多,表型稳定等特征。从遗传资源看,基因组相对较小,易于进行全基因组测序,遗传转化相对容易。从药用特点看,需适于次生代谢产物生物合成和生产研究。
3 本草基因组学的实践应用
本草基因组学作为前沿科学,具有很强的理论性,同时该学科涉及的技术方法和理论对中医药实践具有巨大的指导意义。例如,基于中草药结构基因组开发的DNA条形码分子鉴定技术被国际期刊《生物技术前沿》以题为“草药鉴定从形态到DNA的文艺复兴”发表,将给传统中药鉴定带来革命性影响;基于中草药功能基因组和表观基因组研究阐明道地药材的形成机制,将对优质中药生产和栽培技术的改进提供指导;基于本草基因组学构建的基因数据库、代谢物数据库、蛋白数据库等,以及开发的相关生物信息学方法,将为中药药理学、中药化学、新药开发等提供战略资源;基于合成生物学技术实现目标产物的异源生产,具有环境友好、低耗能、低排放等优点,将为天然药物研发提供全新方式。
3.1 道地药材的生物学本质研究 道地药材是优质药材的代表,既受遗传因素的控制,又受环境条件的影响。组学技术可提供有用工具阐明道地药材的分子机制,例如,道地药材“沙漠人参”肉苁蓉Cistanche deserticola是中国最具特色的干旱区濒危药用植物和关键物种,新疆和内蒙古是其重要主产区和传统道地产区,研究表明,内蒙古阿拉善和新疆北疆是肉苁蓉两大生态适宜生产集中区(2类生态型),黄林芳等[45]对两大产区肉苁蓉化学成分、分子地理标识及生态因子进行考察。应用UPLC-Q-TOF/MS技术对肉苁蓉苯乙醇苷及环烯醚萜苷类成分进行分析;基于psbA-trnH序列对不同产地肉苁蓉进行分子鉴别及分析;通过“中国气象科学数据共享服务网”,获得两大产区包括温度、水分、光照等生态因子数据;运用生物统计、数量分类等分析方法,对肉苁蓉进行生态型划分。UPLC-Q-TOF/MS分析表明,内蒙古与新疆产肉苁蓉明显不同,鉴定出16种成分,其中2′-乙酰毛蕊花糖苷可作为区分两大产地肉苁蓉的指标成分;psbA-trnH序列比对分析发现,肉苁蓉不同产地间序列位点存在差异,新疆产肉苁蓉在191位点为G,内蒙古产则为A,NJ tree分析表明,肉苁蓉2个产地明显分为2支,差异显著;生态因子数据亦表明,肉苁蓉的两大气候地理分布格局,为研究不同生态区域中药生态型及品质变异的生物学本质提供了一种新思路,也为深化道地药材理论研究奠定重要基础。
另外,针对同一药材在不同种植区域,开展中草药表观基因组研究,明确不同生产区域的遗传变异,特别是环境不同对药材表观遗传的修饰作用,包括DNA甲基化修饰、小RNA测序分析、染色质免疫共沉淀分析等。此外,土壤微生物也是道地药材生长环境中的重要因素。采用宏基因组分析土壤微生物群落,为揭示土壤微生物和药材生长的相互作用提供依据。
3.2 中药分子标记用于中药质量控制研究 本草基因组和功能基因组研究为开发药材分子标记提供了丰富基因资源。基于基因组的分子标记有AFLP, ISSR, SNP等,基于转录组的分子标记有SSR等。当前国际上最受关注的分子标记是DNA条形码,已经构建标准操作流程和数据库、鉴定软件,可广泛应用于中药企业、药房、研究院所和大专院校等。中药材DNA条形码分子鉴定指导原则已被纳入《中国药典》,植物药材以ITS2序列为主、psbA-trnH为辅助序列,动物药材以COI序列为主、ITS2为辅助序列,在此基础上,进一步开发了质体基因组作为超级条形码对近缘物种或栽培品种进行鉴定。该体系可广泛应用于中药材种子种苗、中药材、中药超微破壁饮片、中成药等鉴定,已出版专著《中国药典中药材DNA条形码标准序列》和《中药DNA条形码分子鉴定》。
3.3 本草基因资源的保护与利用 随着本草基因组研究的发展,本草遗传信息快速增加,灵芝基因组论文被Nature China网站选为中国最佳研究(图6),迫切需要一个通用平台整合所有组学数据。数个草药数据库已经被建立,例如草药基因组数据库(http://herbalgenomics.org)、转录组数据库(http://medicinalplantgenomics.msu.edu)、草药DNA条形码数据库(http://tcmbarcode.cn/en)、代谢途径数据库(http://cathacyc.org)等。但是这些数据库缺乏长期维护,对使用者要求具备一定生物信息学技能。因此整合DNA和蛋白质序列、代谢组成分信息,方便使用的大数据库十分必要和迫切。进一步提升生物信息分析方法,更好地利用基因组和化学组信息解析次生代谢产物的生物合成途径,将有助于有效设计和寻找植物和真菌药物。
利用简化基因组测序技术获得数以万计的多态性标记。通过高通量测序及信息分析,快速鉴定高标准性的变异标记(SNPs),已广泛应用于分子育种、系统进化、种质资源鉴定等领域。利用该技术可以筛选抗病株的特异SNPs位点,建立筛选三七抗病品种的遗传标记,辅助系统选育,有效的缩短育种年限。通过系统选育的方法获得的抗病群体,并采用RAD-Seq技术筛选抗病株的SNPs位点,为基因组辅助育种提供遗传标记,进而有效缩短了三七的育种年限,加快育种进程。利用遗传图谱识别影响青蒿产量的基因位点取得突破,论文发表于《科学》[7],该文基于转录组及田间表型数据,通过构建遗传图谱识别影响青蒿素产量的位点。青蒿植株表型的变异出现在Artemis的F1谱系中,符合高水平的遗传变异。Graham等[7]发现与青蒿素浓度相关的QTL分别为LG1,LG4及 LG9(位于C4)。在开发标记位点用于育种的同时,Graham等检测了23 000株植株的青蒿素含量,这些植株是青蒿的F1种子经甲基磺酸乙酯诱变后于温室培养12周的F2、F3代。结果发现经诱变后的材料大约每4.5 Mb有一个突变,其变异频率小于Artemis中的每1/104碱基对的SNP多态性。该方法能够识别携带有益变异的个体(来源于甲基磺酸乙酯诱变处理),同时亦能识别遗传背景获得提升的个体(由于自然变异而导致有益等位基因分离的个体)。Graham等也检测高产F2代植株青蒿素的含量:尽管F2的植株杂合性较低,但其青蒿素含量比UK08 F1群体植株的含量高。另外,Graham等验证了基于田间试验获得与青蒿素含量相关的QTL在温室培育的高产植株中高效表达。同时发现,大量分离畸变有利于有益的等位基因(位于C4 LG1且与青蒿素产量相关的QTL)。这些数据证实了QTL及其对青蒿素产量的影响,同时也证明了基因型对于温室及田间培育的青蒿材料具有极大影响。
3.4 中药合成生物学研究 结构复杂多样的中药药用活性成分是中药材发挥药效的物质基础,也是新药发现的重要源泉。然而许多中药材在开发和使用的过程中往往面臨一系列难题,如许多药材生长受环境因素影响较大;有些珍稀药材生长缓慢,甚至难以人工种植;大多数药用活性成分在中药材中含量低微,结构复杂,化学合成困难;传统的天然提取或者人工化学合成的方法难以满足科研和新药研发的需求,中药合成生物学将是解决这一矛盾的有效途径。中药合成生物学是在本草基因组研究基础上,对中药有效成分生物合成相关元器件进行发掘和表征,借助工程学原理对其进行设计和标准化,通过在底盘细胞中装配与集成,重建生物合成途径和代谢网络,实现药用活性成分的定向、高效的异源合成,从而提升我国创新性药物的研发能力和医药产业的国际核心竞争力[40]。
随着基于高通量测序的中草药结构基因组学和转录组学研究的快速发展,利用生物信息学技术和功能基因组学方法从大量中药原物种的遗传信息中筛选和鉴定出特定次生代谢途径的酶编码基因,将极大加快次生代谢途径的解析进程,为中药合成生物学研究奠定坚实基础。通过优化密码子偏好性、提高关键酶编码基因的表达量、下调或抑制代谢支路等方法来优化和改造异源代谢途径, 按人们实际需求获取药用活性成分[40]。
3.5 中药作用靶点与个性化治疗 中药蛋白质组学将蛋白组学技术应用于中药研究领域,对寻找中药的可能靶点和阐明中药有效成分作用机制具有重要意义。譬如,蒋建东教授团队在小檗碱降血脂研究中开展的突出工作[46],以及Pan等[47]利用蛋白组学技术分析丹参酮ⅡA对宫颈癌Caski细胞的抑制作用,发现C/EBP同源蛋白和细胞凋亡信号调节激酶1参与丹参酮ⅡA的抑癌作用。对于中药复方的相关作用靶点也有报道,Nquyen-Khuong等[48]探讨了由栝楼、大豆、中药五味子和西地格丝兰提取物组成的混合物作用于人膀胱癌细胞后蛋白质组的表达谱变化,鉴定了多种与能量代谢、细胞骨架、蛋白质降解以及肿瘤抑制相关的蛋白。
青蒿素及其衍生物青蒿琥酯表现出明显的体内外抗肿瘤活性,但其抗肿瘤的分子机制并不明确。研究者采用了基因芯片技术,在转录水平解析青蒿琥酯抗肿瘤相关的基因。再将表达谱数据导入信号通路分析和转录因子分析,结果表明c-Myc/Max可能是作为肿瘤细胞应对青蒿琥酯效应基因的转录调控因子,这一结果可能指导针对不同个体采用不同的治疗策略[42]。由于银杏具有显著的诱导CYP2C19活性效应,通过研究不同CYP2C19基因型健康中国人个体,银杏与奥美拉唑(omeprazole,广泛使用的CYP2C19底物)潜在的中西药互作关系。结果显示,银杏诱导CYP2C19基因型模式依赖的奥美拉唑羟基化反应,随后降低5-羟基奥美拉唑肾脏清除率。银杏和奥美拉唑或其他CYP2C19底物共同服用可显著减弱其药效,还需更多证据支持[49]。这一研究证实个体化治疗基于人体基因差异,可能发挥更好疗效。
[参考文献]
[1]Sanger F, Air G M, Barrell B G, et al. Nucleotide sequence of bacteriophageφX174 DNA[J]. J Mol Biol, 1978, 125(2):225.
[2]Sachidanandam R, Weissman D, Schmidt S C, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms[J]. Nature, 2001, 409(6822):928.
[3]Venter J C, Adams M D, Sutton G G, et al. Shotgun sequencing of the human genome[J]. Science, 1998, 280(5369):1540.
[4]Initiative A G. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana[J]. Nature, 2000, 408(6814):796.
[5]中国药材公司.中国中药资源 [M]. 北京:科学出版社, 1995.
[6]Chen S, Xu J, Liu C, et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum[J]. Nat Commun, 2012, 3(2):177.
[7]Graham I A, Besser K, Blumer S, et al. The genetic map of Artemisia annua L. identifies loci affecting yield of the anti-malarial drug artemisinin [J]. Science, 2010, 327: 328.
[8]Yan Y, Wang Z, Tian W, et al. Generation and analysis of expressed sequence tags from the medicinal plant Salvia miltiorrhiza[J]. Sci Chin Life Sci, 2010, 53(2):273.
[9]李滢,孙超,罗红梅,等. 基于高通量测序454 GS FLX的丹参转录组学研究[J]. 药学学报,2010, 45(4):524.
[10]Hua W, Zhang Y, Song J, et al. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients [J]. Genomics, 2011, 98(4):272.
[11]Yang L, Ding G, Lin H, et al. Transcriptome analysis of medicinal plant Salvia miltiorrhiza and identification of genes related to tanshinone biosynthesis[J]. PLoS ONE, 2013, 8(11):e80464.
[12]Luo H, Zhu Y, Song J, et al. Transcriptional data mining of Salvia miltiorrhiza in response to methyl jasmonate to examine the mechanism of bioactive compound biosynthesis and regulation[J]. Physiol Plantarum, 2014, 152(2):241.
[13]Ge Q, Zhang Y, Hua W P, et al. Combination of transcriptomic and metabolomic analyses reveals a JAZ repressor in the jasmonate signaling pathway of Salvia miltiorrhiza [J]. Sci Rep, 2015, 5: 14048.
[14]Gao W, Sun H X, Xiao H, et al. Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza [J]. BMC Genomics, 2014, 15:73.
[15]Xu Z, Peters R J, Weirather J, et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza, and tanshinone biosynthesis [J]. Plant J, 2015, 82(6):951.
[16]Sun C, Li Y, Wu Q, et al. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis [J]. BMC Genomics, 2010, 11:262.
[17]Li Y, Luo H M, Sun C, et al. EST analysis reveals putative genes involved in glycyrrhizin biosynthesis [J]. BMC Genomics, 2010, 11: 268.
[18]Zhu Y J, Xu J, Sun C, et al. Chromosome-level genome map provides insights into diverse defense mechanisms in the medicinal fungus Ganoderma sinense [J]. Sci Rep, 2015, 5:11087.
[19]Xu H, Song J Y, Luo H M, et al. Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza[J]. Mol Plant, 2016, 9: 949.
[20]Liang Y, Xiao W, Hui L, et al. The genome of Dendrobium officinale, illuminates the biology of the important traditional Chinese orchid herb [J]. Mol Plant, 2014, 8(6):922.
[21]Zhang G Q, Xu Q, Bian C, et al. The Dendrobium catenatum Lindl.genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution [J]. Sci Rep, 2016, 6: 19029.
[22]陳士林,何柳,刘明珠,等. 本草基因组方法学研究[J].世界科学技术,2010, 12(3):316.
[23]宋经元,罗红梅,李春芳,等. 丹参药用模式植物研究探讨 [J]. 药学学报,2013, 48(7):1099.
[24]Chen S L, Yao H, Han J P, et al. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species [J]. PLoS ONE, 2010, 5(1):e8613.
[25]Pang X H, Song J Y, Zhu Y J, et al. Applying plant DNA barcodes for Rosaceae species identification [J]. Cladistics, 2011, 27: 165.
[26]Dolezel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry [J]. Nat Protoc, 2007, 2(9):2233.
[27]Vu G T, Dear P H, Caligari P D, et al. BAC-HAPPY mapping (BAP mapping): a new and efficient protocol for physical mapping [J]. PLoS ONE, 2010, 5: e9089.
[28]Microbial genetic analysis-OpGen[EB/OL]. [2016-10-16]. http://www.opgen.com/.
[29]Bionano genomics——whole genome mapping with the irys system[EB/OL]. [2016-10-16]. http://bionanogenomics.com/.
[30]Zerbino D R, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs [J]. Genome Res, 2008, 18(5):821.
[31]Chaisson M J, Pevzner P A. Short read fragment assembly of bacterial genomes [J]. Genome Res, 2008, 18(2):324.
[32]Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo, assembler [J]. Gigascience, 2012, 1(1):18.
[33]Huang X, Madan A. CAP3: a DNA sequence assembly program [J]. Genome Res, 1999, 9(9):868.
[34]Lynn A M, Jain C K, Kosalai K, et al. An automated annotation tool for genomic DNA sequences using GeneScan and BLAST [J]. J Genet, 2001, 80: 9
[35]Solovyev V, Kosarev P, Seledsov I, et al. Automatic annotation of eukaryotic genes, pseudogenes and promoters [J]. Genome Biol, 2006, 7(Suppl 1): S10.
[36]Zdobnov E M, Apweiler R. InterProScan——an integration platform for the signature recognition methods in InterPro [J]. Bioinformatics, 2001, 17(9):847.
[37]Joslyn C A, Mniszewski S M, Fulmer A, et al. The gene ontology categorizer [J]. Bioinformatics, 2004, 20(1):169.
[38]Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG [J]. Nucleic Acids Res, 2006, 34:354.
[39]陳士林,孙永珍,徐江,等. 本草基因组计划研究策略[J]. 药学学报, 2010, 45 (7), 807.
[40]陈士林, 朱孝轩, 李春芳, 等. 中药基因组学与合成生物学[J].药学学报,2012, 47 (8): 1070.
[41]Chen S L, Song J Y, Sun C, et al. Herbal genomics: examining the biology of traditional medicines [J]. Science, 2015, 347 (6219 Suppl): S27.
[42]Sertel S, Eichhorn T, Simon C H, et al.Pharmacogenomic identification of c-Myc/Max-regulated genes associated with cytotoxicity of artesunate towards human colon, ovarian and lung cancer cell lines [J]. Molecules, 2010, 15(4): 2886.
[43]Scherf U, Ross D T, Waltham M, et al.A gene expression database for the molecular pharmacology of cancer [J]. Nat Genet, 2000, 24(3): 236.
[44]Chen Y, Ouyang D S, Kang Z, et al. Effect of a traditional Chinese medicine Liu Wei Di Huang Wan on the activities of CYP2C19, CYP2D6 and CYP3A4 in healthy volunteers [J]. Xenobiotica, 2012, 42(6): 596.
[45]黃林芳,郑司浩,武拉斌,等. 基于化学成分及分子特征中药材肉苁蓉生态型研究 [J]. 中国科学: 生命科学, 2014, 44(3): 318.
[46]Kong W, Wei J, Abidi P, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins [J].Nat Med, 2004, 10(12):1344.
[47]Pan T L, Wang P W, Hung Y C, et al. Proteomic analysis reveals tanshinone ⅡA enhances apoptosis of advanced cervix carcinoma CaSki cells through mitochondria intrinsic and endoplasmic reticulum stress pathways [J].Proteomics, 2013, 13(23/24):3411.
[48]Nguyen-Khuong T, White M Y, Hung T T, et al. Alterations to the protein profile of bladder carcinoma cell lines induced by plant extract MINA-05 in vitro [J].Proteomics, 2009, 9(7):1883.
[49]Yin O Q, Tomlinson B, Waye M M, et al. Pharmacogenetics and herb-drug interactions: experience with Ginkgo biloba and omeprazole[J].Pharmacogenetics, 2004, 14(12):841.
[责任编辑 丁广治]
猜你喜欢
中老年保健(2021年4期)2021-12-01
中老年保健(2021年4期)2021-08-22
成都医学院学报(2021年2期)2021-07-19
科学(2020年2期)2020-08-24
金桥(2020年7期)2020-08-13
国际口腔医学杂志(2019年3期)2019-05-31
基层中医药(2018年6期)2018-08-29
天然产物研究与开发(2018年2期)2018-04-04
医学研究杂志(2015年11期)2015-06-10
生物进化(2014年3期)2014-04-16
