
在线固相萃取-同位素稀释/超高效液相色谱-串联质谱法测定蜂蜜中26种喹诺酮类化合物
2021-11-03 09:17张居舟
分析测试学报 2021年10期
张居舟,李 静
(1. 安徽省食品药品检验研究院,安徽 合肥 230051;2. 国家农副加工食品质量检验检测中心,安徽 合肥 230051)
喹诺酮类药物为人工合成的含4-喹诺酮基本结构的广谱抗生素,可作用于细菌的脱氧核糖核酸的回旋酶,使细胞无法分裂,从而起到抗菌效果[1]。该类药物目前已发展到第五代,为人畜共用,可预防不同种类动物的疾病。喹诺酮类药物可防治蜜蜂的腐臭病、败血病等,然而药物滥用致使其在蜂产品中残留。人类若长期食用含有喹诺酮类药物的食品,会诱导致病菌产生耐药性;若过量食用此类药物,还会出现恶心、腹泻、头痛、眩晕,以及白细胞减少、肝损伤等不良反应,有致畸、致突变的潜在风险[2-3]。因此,国内外对食品中喹诺酮的含量均进行严格限定。我国规定,恩诺沙星、达氟沙星、双氟沙星和沙拉沙星在家禽产蛋期禁用,并规定了其他动物源食品中的最大残留限量[4];洛美沙星、培氟沙星、氧氟沙星和诺氟沙星在食品动物中禁用[5]。欧盟规定了麻保沙星、环丙沙星、达氟沙星、恩诺沙星、沙拉沙星、双氟沙星、氟甲喹和噁喹酸的最大残留限量,此类抗生素药物也未被授权用于治疗蜜蜂[6]。为规避市场监管,仍有企业非法使用标准规定之外的喹诺酮类抗生素,然而新一代喹诺酮类药物的检测方法尚未建立。因此,迫切需要开发一种能同时测定蜂蜜中目前上市的所有喹诺酮类药物的检测方法,进而为监管部门及蜂蜜进出口贸易提供技术支持。
目前,蜂蜜中喹诺酮类药物的检测方法有薄层色谱法[7]、高效液相色谱法(HPLC)[8-9]、高效液相色谱-质谱/质谱法(HPLC-MS/MS)[10-16]等,其中薄层色谱法适用于定性分析,HPLC 法的定性易受假阳性干扰,HPLC-MS/MS法集多组分的定性、定量和高效分离于一体,应用最为广泛。喹诺酮类药物结构相似,且存在同分异构体,文献报道的前处理方法有经典固相萃取[10,14]、分子印迹固相萃取[16]、磁性多壁碳纳米管固相萃取[15]、光纤固相微萃取[11]和QuEChERS[8,12]等,然而针对蜂蜜基质,上述方法均有各自的优势和不足。本文应用在线固相萃取技术,提升了前处理效率,采用超高效液相色谱-串联质谱的动态多反应监测(dMRM)模式,系统自动分配扫描时间,驻留时间更长,明显提高了灵敏度,适合高通量快速分析;检测26种目标物,较文献[10]和国标[17-18]方法新增了尚未报道的那氟沙星、妥苏沙星、克林沙星、莫西沙星、吉米沙星和巴洛沙星6 种药物,可以满足蜂蜜中喹诺酮类化合物高通量快速筛查和食品安全应急检验的需要。
1 实验部分
1.1 仪器、试剂与材料
Agilent 1290 超高效液相色谱仪、6495 型三重四极杆串联质谱仪(配电喷雾离子源,美国Agilent 公司),GX-271 ASPEC 全自动固相萃取仪(美国Gilson 公司),IKA MS3 Basic 型涡旋振荡器(i国IKA 公司)。Oasis PRiME HLB 固相萃取柱(200 mg/6 mL,美国Waters公司)。
标准品:克林沙星标准溶液(100 μg/mL,美国A ChemTek 公司);加替沙星(纯度98.5%)、盐酸莫西沙星(纯度99.8%)购于美国CATO 公司;吉米沙星(纯度97%)、巴洛沙星(纯度98%)、盐酸妥苏沙星(纯度97%)购于加拿大TRC 公司;那氟沙星(纯度97.6%)购于i国Dr. Ehrenstorfer 公司;其余19 种喹诺酮类混合标准溶液(100 μg/mL)购于美国A ChemTek 公司。内标:恩诺沙星-D5、诺氟沙星-D5、氧氟沙星-D3(i国Dr. Ehrenstorfer 公司);环丙沙星-D8、培氟沙星-D5、沙拉沙星-D8、依诺沙星-D8、双氟沙星-D3购于北京Manhage生物科技公司;氟罗沙星-D3、洛美沙星-D5、达氟沙星-D3、氟甲喹-13C3(美国A ChemTek公司),加替沙星-D4、莫西沙星-13CD3购于加拿大TRC公司,噁喹酸-D5购于上海ANPEL公司。
甲醇、乙腈(LC-MS 级)购于i国Merck 公司;甲酸(色谱纯)购于美国ACS 公司;实验用水由Milli-Q超纯水机制备。
样品:洋槐蜜、枣花蜜、荆条蜜、油菜花蜜、紫云英蜜、枸杞蜜、枇杷蜜、百花蜜、樱花蜜均为本地市售。
1.2 实验方法
1.2.1 标准溶液的配制 混合标准溶液:分别准确称取10.0 mg(精确至0.01 mg)加替沙星、莫西沙星、吉米沙星、巴洛沙星、妥苏沙星、那氟沙星固体标准品至100 mL容量瓶,用甲醇溶解并定容至刻度,配成质量浓度为100 μg/mL的6种混合标准储备液。再分别吸取1 mL克林沙星标准溶液、6种混合标准储备液、其余19 种喹诺酮类药物混合标准溶液于100 mL 容量瓶中,配成质量浓度为1 μg/mL 的26种混合标准溶液,于-18 ℃下避光储存。
同位素内标混合溶液:使用甲醇-水溶液(体积比3∶1,诺氟沙星-D5、培氟沙星-D5溶液分别加2 滴盐酸助溶)分别配制100 μg/mL 的各单标储备液。再用甲醇稀释成1 μg/mL 的15 种混合内标溶液,于-18 ℃下避光储存。
分别精密量取上述混合标准溶液及同位素内标混合溶液适量,用1%甲酸溶液-甲醇(体积比10∶90)稀释成0.1、0.2、0.4、1.0、2.0、5.0、10、20 μg/L 的系列标准工作液,其中内标的质量浓度均为10 μg/L。
1.2.2 样品前处理 称取2 g(精确至0.001 g)蜂蜜于50 mL离心管中,准确加入100 μL 1 mg/L混合内标溶液和10.0 mL 1%甲酸溶液-甲醇(10∶90),涡旋混合至样品溶解,10 000 r/min 离心3 min。将PRiME HLB 小柱置于全自动固相萃取仪萃取架上,调节流速为1 mL/min,吸取2 mL提取液过柱,收集流出液,过0.22 μm滤膜至样品瓶,待分析。
1.2.3 色谱参数 色谱柱:Agilent Eclipse Plus RRHD C18(2.1 mm×50 mm,1.8 μm),流动相:A 为0.1%甲酸溶液,B 为乙腈;线性梯度洗脱程序:0~1.0 min,95% A;1.0~2.0 min,95%~90% A;2.0~3.0 min,90%~87% A;3.0~5.0 min,87%~85% A;5.0~7.0 min,85%~35% A;7.0~8.0 min,35%~10% A;8.0~9.0 min,10% A;9.0~11.0 min,10%~95% A。流速:0.3 mL/min;柱温:35 ℃;进样体积:2 μL。
1.2.4 质谱参数 离子源:电喷雾离子源(AJS ESI);监测方式:dMRM;扫描模式:正离子扫描;干燥气温度:220 ℃;干燥气流速:17 L/min;雾化气压力:2.1×105Pa;鞘气温度:340 ℃;鞘气流速:11 L/min;毛细管电压:3 000 V;喷嘴电压:400 V;碰撞池加速电压:5 V;碎裂电压:380 V;其它质谱参数见表1。

表1 26种喹诺酮类化合物及15种内标的质谱参数Table 1 MS/MS parameters of the 26 quinolones and 15 internal standards
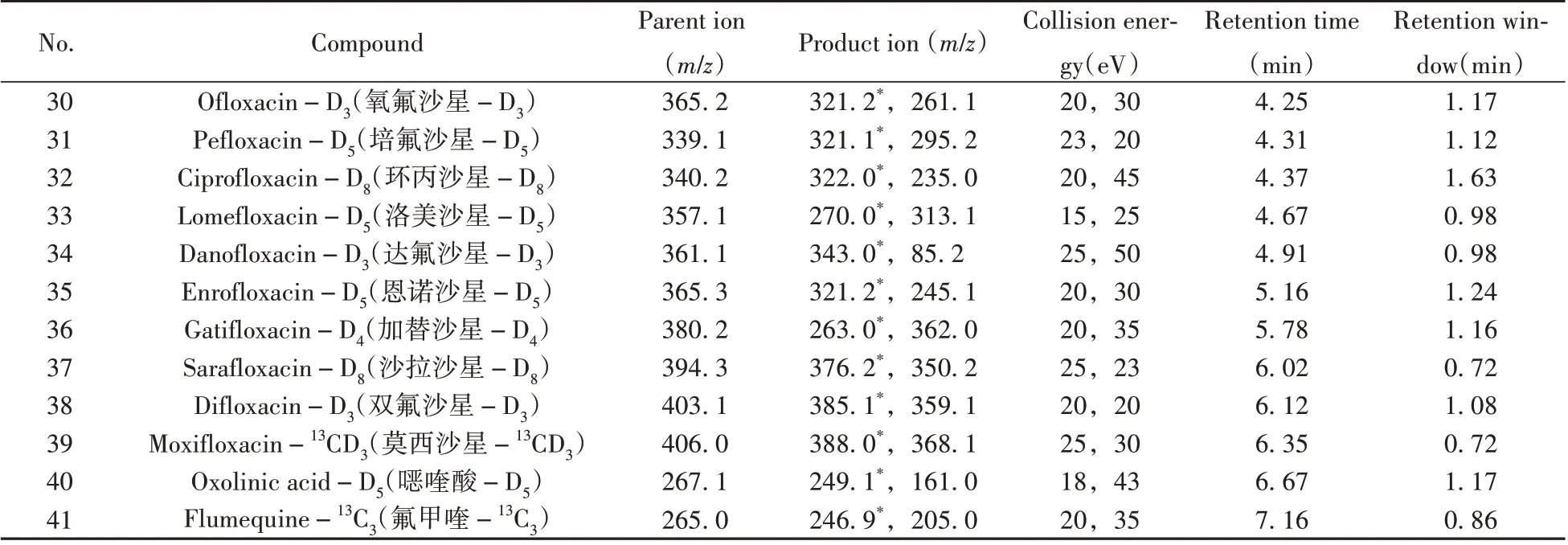
(续表1)
2 结果与讨论
2.1 质谱条件的优化
喹诺酮类化合物结构含有哌嗪环、羧基或喹啉环,其[M+H]+峰响应强度均较大,因此选择[M+H]+作为分子离子峰。使用电喷雾离子源,正离子模式(ESI+),在m/z100~500 范围内对母离子进行全扫描,比较各化合物的母离子响应强度,依次优化毛细管电压、干燥气温度、干燥气流速、雾化气压力、鞘气温度、鞘气流速和喷嘴电压;再分别进行子离子扫描,子离子主要为脱羧峰[M +H-CO2]+和脱羧后哌嗪环断裂重排峰[M+H-CO2-C2H4NR]+[10],在优化碰撞能及碰撞池加速电压后,形成多反应监测(MRM)方法,进一步优化色谱条件,进行数据采集,在MRM 方法数据或图谱的基础上升级形成dMRM方法,每个化合物的保留时间窗口自动进行动态分配,灵敏度和峰形均有不同程度的改善。值得注意的是,氟甲喹和噁喹酸、恩诺沙星-D5和氧氟沙星-D3互为同分异构体,具有相同的母离子和碎片离子信息,需分别优化其单标标准溶液以确定各离子对的碰撞能,并通过各同分异构体在色谱柱上保留时间的差异,设置保留时间窗对峰进行定位。
2.2 色谱条件的优化
喹诺酮类化合物结构含有亲油亲水两性基团,极性存在一定差异,实验分别选择Agilent Eclipse Plus RRHD C18(2.1 mm×50 mm,1.8 μm)、Agilent Eclipse Plus RRHD C18(2.1 mm×100 mm,1.8 μm)和Waters BEH HILIC(2.1 mm×100 mm,1.7 μm)色谱柱进行分析,为避免在反向色谱柱上出现色谱峰拖尾现象,选择0.1%甲酸溶液-乙腈为流动相,依次优化流动相的初始比、梯度洗脱曲线斜率和洗脱时间。结果表明,0.1%甲酸溶液不仅可为正离子扫描提供质子,提高离子化效率,且提高了峰的对称性;C18色谱柱较HILIC 色谱柱的分离效果更佳。随着洗脱曲线斜率的变小,洗脱时间的延长,各化合物被洗脱的速率变缓,可将互为同分异构体的氟甲喹和噁喹酸、恩诺沙星-D5和氧氟沙星-D3完全分离,26 种化合物的灵敏度和分辨率均满足要求。在优化的梯度洗脱程序下,由于100 mm 长度的C18和HILIC 色谱柱需更长的分离时间,且分离效果和峰灵敏度均比50 mm 长度的色谱柱差,因此实验采用50 mm的Agilent Eclipse Plus RRHD C18(2.1 mm×50 mm,1.8 μm)色谱柱,目标化合物的分离效果良好(如图1)。
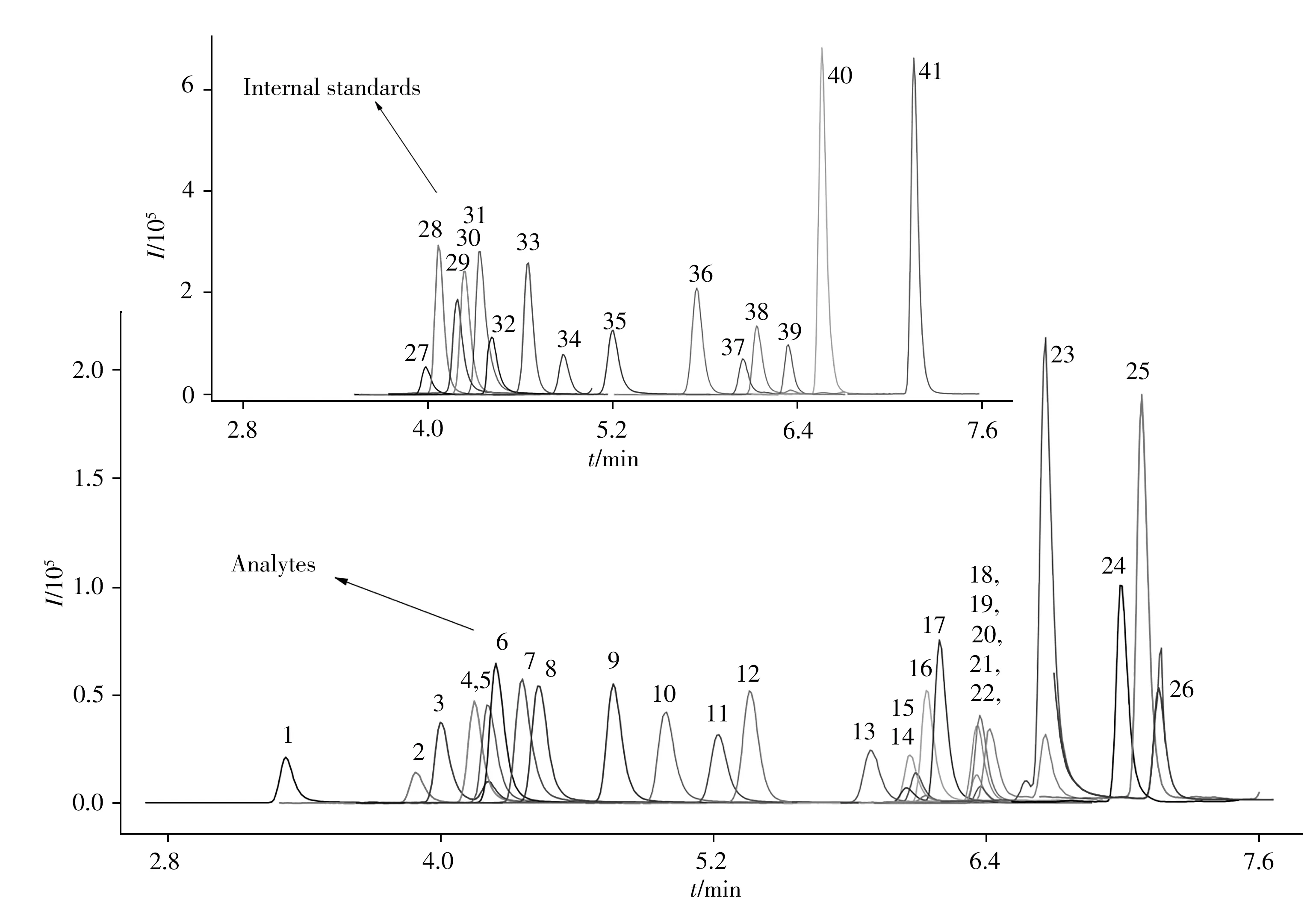
图1 26种喹诺酮类化合物(2.0 μg/L)及15种内标(10 μg/L)的总离子流图Fig.1 Total ion chromatograms of 26 quinolones(2.0 μg/L)and 15 internal standards(10 μg/L)the peak numbers denoted were the same as those in Table 1
2.3 提取条件的选择
喹诺酮类化合物中含有叔氨基和羧基,溶于碱性或酸性溶液,可使用酸性或碱性溶液混合有机溶剂提取。实验对1%甲酸溶液-乙腈(5∶95)、1%甲酸溶液-甲醇(5∶95)、氨水-甲醇(5∶95)的提取效果进行了比较。结果表明,1%甲酸溶液-乙腈(5∶95)的提取液有少量白色沉淀,26 种待测物的回收率为65%~115%,其中氟罗沙星、克林沙星的回收率低于70%;1%甲酸溶液-甲醇(5∶95)可将蜂蜜样品完全溶解,26 种待测物的回收率为70%~128%,其中双氟沙星的回收率≥120%。氨水-甲醇(5∶95)也能完全溶解样品,除奥比沙星的回收率为32%外,其它待测物的回收率为75%~125%。
为进一步验证各提取剂的效果,取6组阳性蜂蜜(其中含有不同浓度的诺氟沙星、氧氟沙星、恩诺沙星及环丙沙星),应用上述3种提取剂进行提取。结果显示,1%甲酸溶液-乙腈(5∶95)提取液只检出氧氟沙星;氨水-甲醇(5∶95)提取液对低浓度样品未检出,1%甲酸溶液-甲醇(5∶95)提取液与国标GB/T 20757-2006[17]的检测结果基本一致,故选择1%甲酸溶液-甲醇(5∶95)作为提取剂,并比较了不同体积比(50∶50、40∶60、30∶70、20∶80、10∶90、5∶95)的1%甲酸溶液-甲醇对26种待测物的回收率(如图2)。结果显示,体积比为20∶80时,除吡派酸、氟罗沙星、氧氟沙星、洛美沙星外,其余化合物的回收率明显偏离100%,体积比为50∶50时整体回收率降至最低,净化时的流速也最慢。实验选择对26种待测物回收率更佳的1%甲酸溶液-甲醇(10∶90)作为提取剂。
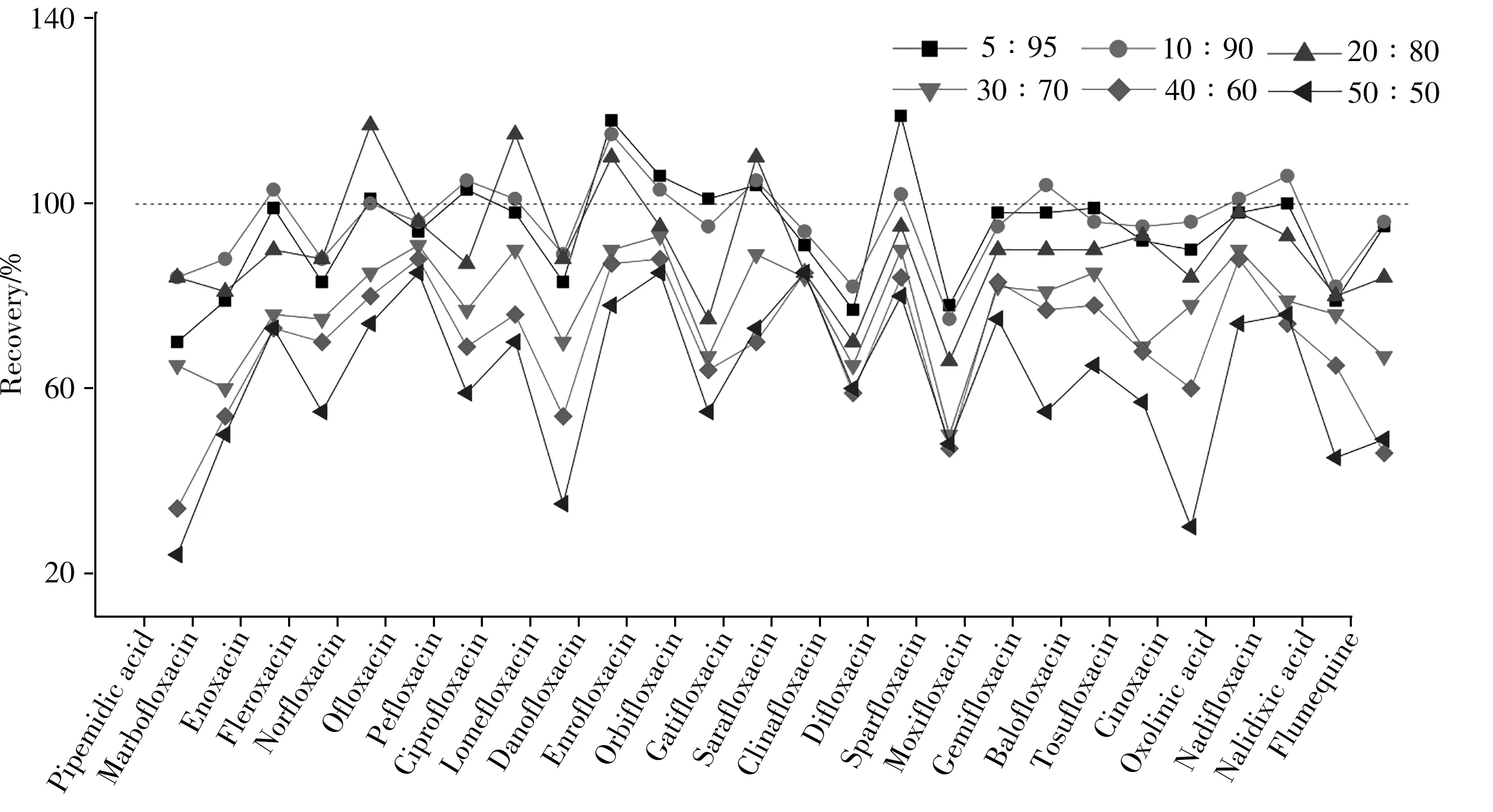
图2 不同体积比的1%甲酸溶液-甲醇对26种待测物的提取效果Fig.2 Extraction efficiencies of 26 analytes by different volume ratios of 1% formic acid solution-methanol
2.4 净化条件的选择
蜂蜜含有氨基酸、有机酸、糖类、色素和少量蛋白质等[19],针对蜂蜜的净化方法有固相萃取和QuEChERS 等方法,本文考察了PRiME HLB 柱、HLB 柱[17]、PAX 柱[18]和QuEChERS[12]的净化效果。称取空白基质样品进行10.0 μg/kg 的加标回收实验,结果表明,PRiME HLB 柱可去除蜂蜜中有机酸、部分色素及糖类等杂质。从去除杂质效果看,HLB 柱与PAX 柱相当,PRiME HLB 柱和QuEChERS 次之。从回收率上看,PRiME HLB 柱优于HLB柱,均有24种目标物的回收率在80%~110%之间;QuEChERS次之;PAX 最差,只有10 种化合物的回收率大于80%,其余在22%~78%之间。实验最终选择净化能力略弱,但回收率较好,且无需活化、淋洗和洗脱,操作过程更简便的PRiME HLB柱进行净化。
2.5 基质效应评价
基质效应(ME)是指在测定过程中,由于待测化合物的离子化效应被样品基质改变,从而使响应信号受到增强或抑制的现象。目前评价基质效应的方法有标准曲线法和相对响应值法,本文采用标准曲线法考察喹诺酮类化合物在蜂蜜中的基质效应,ME=基质匹配标准曲线斜率/溶剂标准曲线斜率×100%。当ME>115%时为基质增强效应;当85%≤ME ≤115%时基质效应可忽略;当ME<85%时为基质抑制效应[20-21]。实验考察了洋槐蜜、油菜花蜜和荆条蜜的基质效应(分别以ME1、ME2、ME3表示),表2结果表明,目标化合物及其同位素内标在各个峰蜜中的基质效应变化趋势一致,荆条蜜较其他蜜的基质效应略小,奥比沙星、加替沙星等8种化合物的基质效应不明显(89%~115%),麻保沙星等18种化合物为基质增强效应,其中,吡派酸、依诺沙星、诺氟沙星、培氟沙星、达氟沙星、环丙沙星的基质增强效应较强。
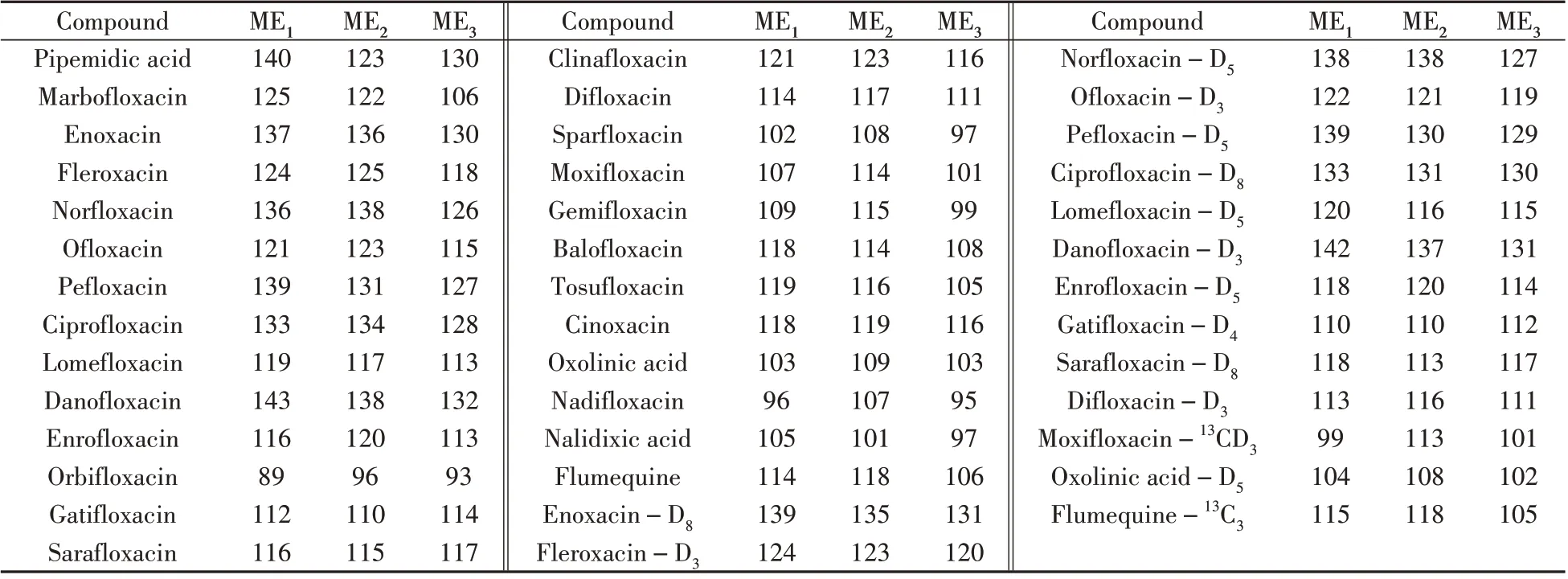
表2 26种喹诺酮类化合物及15种内标的基质效应(%)Table 2 Matrix effects of 26 quinolones and 15 internal standards(%)
基质效应的消除方法通常有基质匹配曲线、同位素内标、基质净化法等[22],本文选择内标法减弱基质效应对目标物定量结果的影响。对于有同位素内标的待测物,选择其同位素内标作为内标定量;对于无同位素内标的化合物,依据结构相似、保留时间相近、基质效应差异较小的原则选择其它同位素内标定量(见表3)。
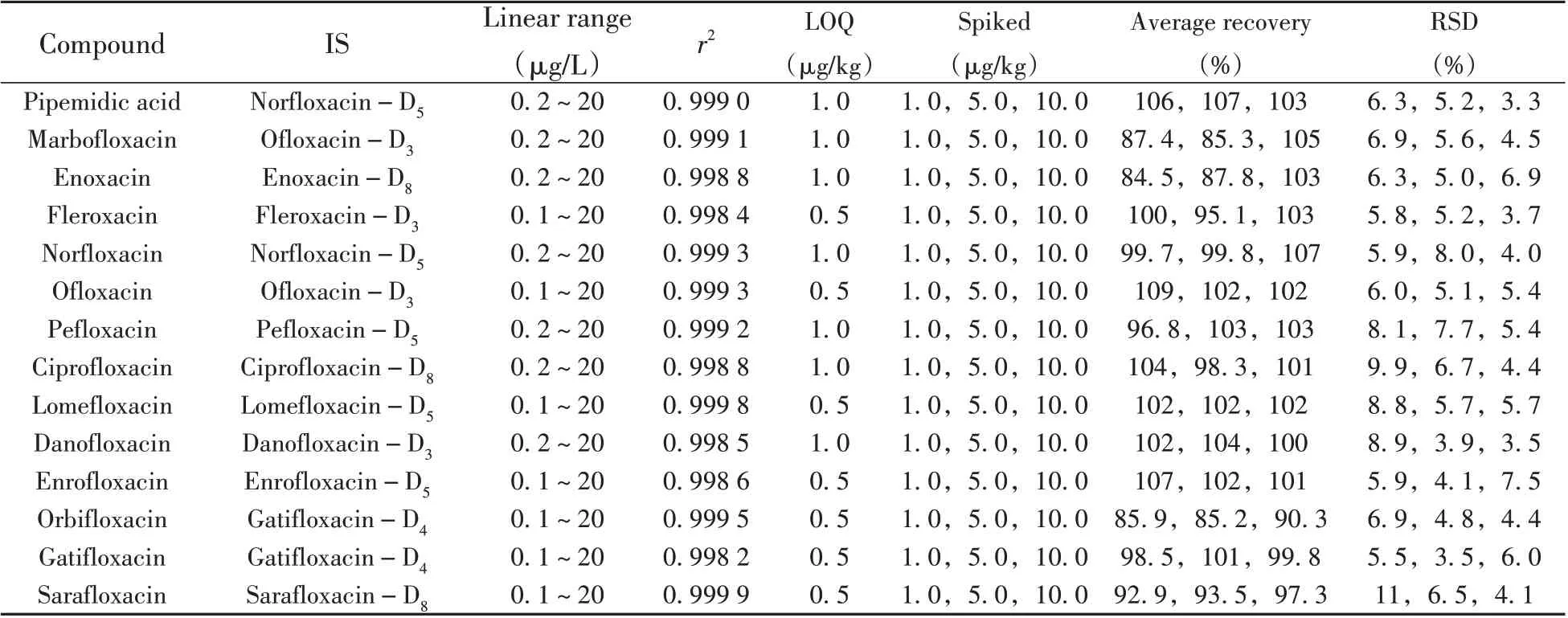
表3 26种喹诺酮类化合物的线性范围、相关系数、定量下限、平均回收率及相对标准偏差(n=6)Table 3 Linear ranges,correlation coefficients(r2),limits of quantitation,average recoveries and relative standard deviations of 26 quinolones(n=6)
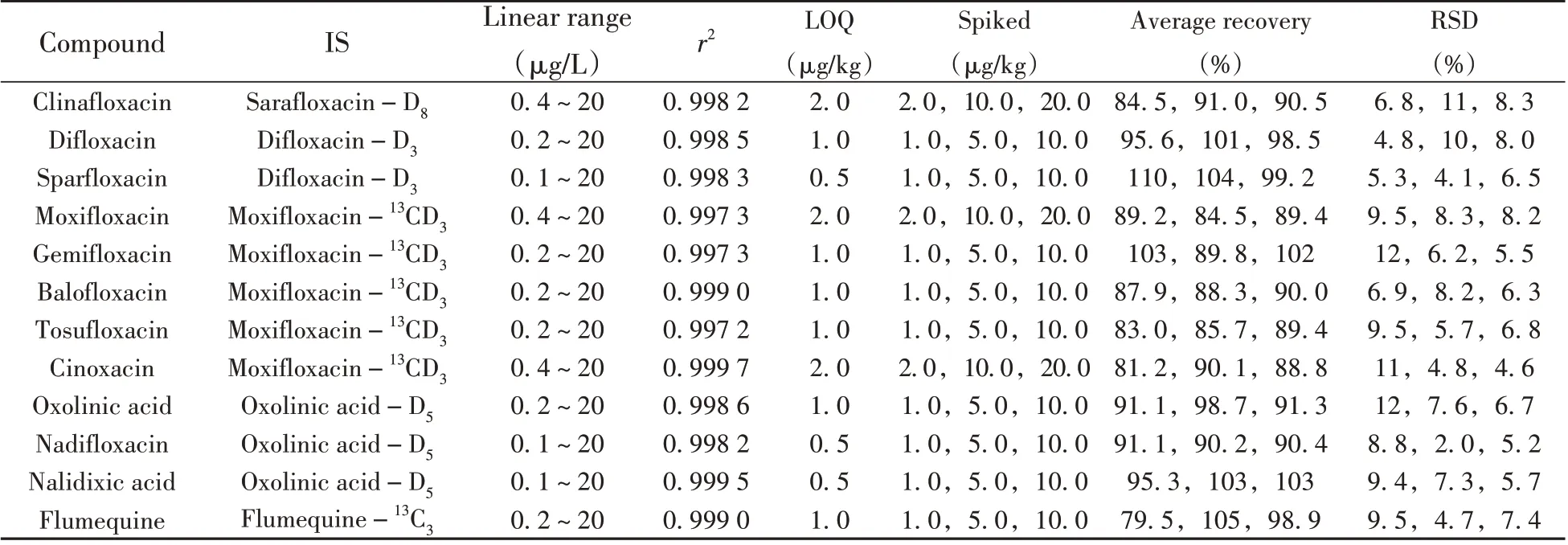
(续表3)
2.6 线性范围与定量下限
精密量取适量喹诺酮类化合物混合标准溶液,用初始流动相配制质量浓度为0.1~20 μg/L 的系列标准工作溶液,同时加入15 种内标(内标质量浓度均为10 μg/L),按优化的条件进行UPLC-MS/MS 分析。以定量离子和内标化合物的峰面积比为纵坐标(y),待测化合物和内标化合物的质量浓度比为横坐标(x),绘制标准工作曲线,内标法定量。结果表明,26 种喹诺酮类化合物在各自的质量浓度范围内线性良好,相关系数(r2)>0.997。采用空白蜂蜜样品添加待测物,以10倍信噪比(S/N≥10)对应的质量浓度确定为定量下限(LOQ),结果表明,26种喹诺酮类化合物的LOQ为0.5~2.0 μg/kg(见表3)。
2.7 准确度与精密度
按照本方法对空白洋槐蜜进行加标回收实验,加标水平为低、中、高3 个浓度,每个浓度平行6 个。由表3 可知,26 种喹诺酮类化合物的平均回收率为79.5%~110%,相对标准偏差(RSD)为2.0%~12%,表明方法的准确度和精密度良好。
2.8 实际样品检测
为评价该方法的有效性,本实验测定了随机抽取的市售洋槐蜜、枣花蜜、荆条蜜、油菜花蜜、紫云英蜜各10份,枸杞蜜、枇杷蜜各5份,百花蜜、樱花蜜各1份,共62份样品。在2份紫云英蜜、3份洋槐蜜样品中检出诺氟沙星,检出量为2.1~22.1 μg/kg;1份枣花蜜样品中检出氧氟沙星,检出量为17.1 μg/kg;1份枇杷蜜中同时检出3.4 μg/kg诺氟沙星和8.8 μg/kg氧氟沙星(如图3);其余样品均未检出目标化合物。
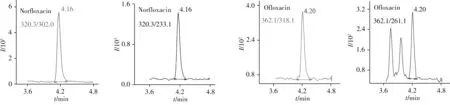
图3 阳性样品中诺氟沙星和氧氟沙星的选择离子色谱图Fig.3 Selected ion chromatograms of norfloxacin and ofloxacin in positive sample
3 结 论
本文建立了一种同时检测蜂蜜中26种喹诺酮类药物的在线固相萃取/超高效液相色谱-串联质谱方法。蜂蜜样品以1%甲酸溶液-甲醇(10∶90)提取,经PRiME HLB 小柱在线通过式固相萃取净化后,回收率优于标准方法;液质联用仪采用dMRM 采集模式,进一步提升了检测灵敏度;15种同位素内标有效补偿了基质效应带来的定量偏差;并首次检测了巴洛沙星等新一代喹诺酮类药物,填补了相关物质检测技术的空白。本方法操作简便、快速,重复性好、灵敏度高、准确可靠,完全满足蜂蜜的实际检验和风险监测需要。
猜你喜欢
中国合理用药探索(2022年3期)2022-11-25
分子催化(2022年1期)2022-11-02
中草药(2022年9期)2022-05-06
中国药学药品知识仓库(2022年1期)2022-03-23
中国药学药品知识仓库(2022年2期)2022-03-23
医药导报(2021年8期)2021-11-30
系统医学(2021年10期)2021-08-04
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
农民致富之友(2018年12期)2018-06-29
湖北农业科学(2017年24期)2018-01-27
