
QuEChERS结合HPLC-MS/MS同时测定鱼肉中多种兽药残留
2021-10-22 00:22刘进玺王铁良胡京枝杨亚琴冯慧慧钟红舰
食品与发酵工业 2021年19期
刘进玺,王铁良,胡京枝,杨亚琴,冯慧慧,钟红舰
(河南省农业科学院 农业质量标准与检测技术研究所,农业部农产品质量监督检验测试中心,农业部农产品质量安全风险评估重点实验室,河南 郑州,450002)
鱼肉是居民餐桌上的重要组成部分,其安全性与人民生活息息相关。随着现代水产养殖业呈集约化和规模化发展,水产生物疫情的爆发频率也逐渐增高,而生产者和经营者为了预防和治疗各种病害、提高产量效益,常在水产生物中使用药物,加上对这些药物的毒性和危害认识不足,导致超量和超种类的滥用、误用、违规使用,以及不遵守休药期等现象屡禁不止,水产品中药物残留超标现象时有发生[1-4]。磺胺类、喹诺酮类、酰胺醇类、硝基咪唑类药物以及一些违禁添加物,例如孔雀石绿等,因为价格低廉,药效好而被经常使用。这些残留在水产品中的药物经食用后在人体中蓄积,会产生致畸、致癌等严重危害[5]。因此,如何提高检验效率、降低检测成本,控制和消除药残,保证食品安全,是重大民生课题[6-7]。
由于不同种类间药物的化学性质差别较大,兽药多残留检测存在诸多困难,如残留量甚微、兽药品种繁多、动物源性食品基质复杂等,目前常用的检测方法大多是测定某一种或某一类药物。然而在实际工作当中,对于同一个样品测定多种药物残留时,分别测定会涉及多个样品前处理和多种类型的仪器,耗费大量的人力、物力和时间,检验成本高且对环境不友好,面对一些突发事件和应急任务时,分别测定的方法就显得力不从心,因此,建立简便、快速、高通量、高灵敏度的方法已成为未来兽药残留检测的趋势[8-10]。液相色谱串联质谱技术集中了色谱和质谱的优点,将色谱的分离能力和质谱的高选择性、高灵敏度结合起来,是近几年来水、土、动物源性食品及排泄物等基质中多类兽药残留快速测定的首选[4,11-21]。
2019年,国家食品安全监督抽查针对淡水鱼类共涵盖了9类药物残留,包括喹诺酮类、磺胺类、酰胺醇类、硝基呋喃类代谢物、硝基咪唑类、四环素族、地西泮、五氯酚酸钠和孔雀石绿,涉及的检测方法除磺胺类和喹诺酮类药物可以用《农业部1077号公告-1-2008》同时测定外,其余7类药物残留均采用国标或相应标准中检测方法(GB/T 19857—2005《水产品中孔雀石绿和结晶紫残留量的测定》、GB/T 20756—2006《可食动物肌肉、肝脏和水产品中氯霉素、甲砜霉素和氟苯尼考残留量的测定 液相色谱-串联质谱法》、SN/T 3235—2012《出口动物源食品中多类禁用药物残留量检测方法 液相色谱-质谱/质谱法》、GB/T 21318—2007《动物源性食品中硝基咪唑残留量检测方法》、GB 23200.92—2016《食品安全国家标准 动物源性食品中五氯酚残留量的测定 液相色谱-质谱法》)。针对这种情况,本文结合2019年的监督抽查任务,将鱼肉中的6类药物残留使用QuEChERS(quick,easy,cheap,effective,rugged,safe)的方法进行前处理,利用高效液相色谱串联质谱(LC-MS/MS)技术,建立了一种同时测定鱼肉中磺胺类、喹诺酮类、酰胺醇类、硝基咪唑类、地西泮和孔雀石绿类药物残留的检测技术。
1 材料与方法
1.1 仪器与试剂
仪器设备:HPLC-MS/MS 8050液相色谱串联质谱仪,日本Shimadzu公司;Milli-Q超纯水发生器,美国Millipore公司;GL-21B高速冷冻离心机,上海安亭科学仪器厂;雷磁PH S-3C酸度计,上海天美科学仪器有限公司;均质仪,美国Tomtec公司。
试剂:乙腈、甲醇、丙酮,均为色谱纯,德国Merck;Na2SO4、NaCl、盐酸、MgSO4,均为分析纯,国药集团化学试剂有限公司;兽药标准品,德国Dr.Ehrenstorfer GmbH。
1.2 标准溶液的配制
准确称取10 mg(精确至0.01 mg)各个兽药标准品于10 mL容量瓶中,根据各兽药的性质用乙腈、甲醇、丙酮等溶剂溶解并定容,制得25种兽药的单一标准储备液(1 mg/mL)。根据需要用乙腈稀释成相应浓度的单一标准工作液。分别吸取一定量的标准储备液,配制成混合标准储备液,临用前稀释成合适浓度的混合标准工作液。
1.3 HPLC-MS/MS条件
液相色谱条件:CAPCELL PAK C18色谱柱(2.0 mm×100 mm×3 μm),日本资生堂公司;柱温40 ℃;流动相A相为5 mmol/L甲酸-甲酸铵水溶液,B相为V(乙腈)∶V(甲醇)=1∶1;线性梯度洗脱程序:0~2 min,90% A,2~3 min,90%~85% A,保持10 min,13~20 min,85%~10% A,保持5 min,25~25.1 min,85%~10%A,保持至28 min;流速0.2 mL/min;进样体积1 μL。
质谱测定条件:电喷雾离子源;正负离子同时扫描;多反应监测模式;各个分析物的质谱信息见表1。
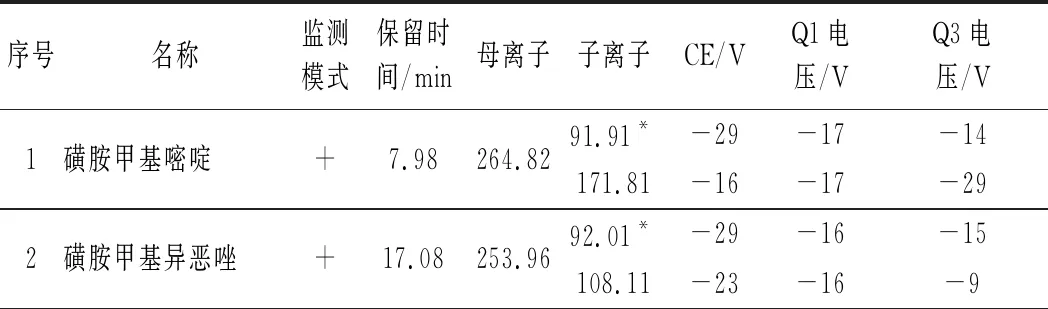
表1 25种分析物的质谱采集参数及保留时间Table 1 MS/MS parameters and retention times of 25 analytes
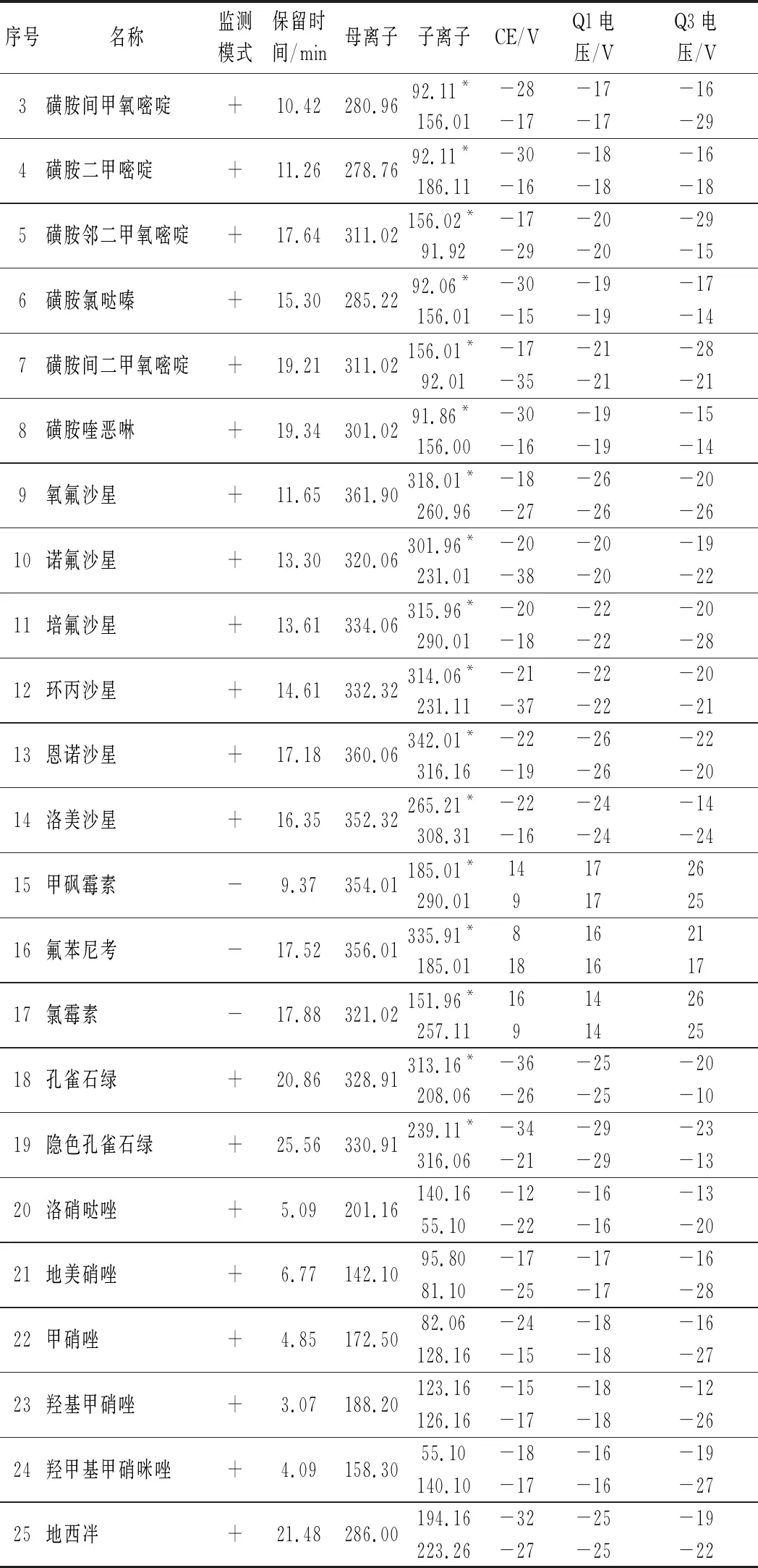
续表1
1.4 样品前处理
精确称量5 g样品于50 mL离心管中,加入4 mL 0.1 mol/L Na2EDTA-Mcllvaine缓冲溶液(pH 4.0)和16 mL 5%甲酸乙腈,剧烈振揺1 min;加入NaCl(1 g)和Na2SO4(4 g),高速均质分散3 min,然后涡旋2 min;10 000 r/min离心12 min备用。
取一个10 mL离心管,装入300 mg MgSO4和50 mg C18,然后吸取2 mL样品上清液,涡旋30 s,然后以6 000 r/min离心5 min。吸取1 mL上清液于另一干净离心管中,加入1 mL水,混匀,过0.22 μm滤膜,待检测。
2 结果与讨论
2.1 仪器条件的选择
2.1.1 液相条件的确认
水相的选择方面,实验对比了加入甲酸、甲酸铵、乙酸、乙酸铵以及它们不同浓度组合时的分离效果,发现加入5 mmol/L甲酸铵-甲酸水溶液时各个分析物的峰形和分离效果最好。有机相的选择方面,实验尝试了乙腈、甲醇以及乙腈和甲醇不同比例组合时的分析效果,结果表明当乙腈和甲醇比例为1∶1时,综合分离度和峰形最优。综合考虑最终确定以V(5 mmol/L甲酸铵-甲酸水溶液)∶V(乙腈-甲醇)=1∶1作为流动相。
为了获得更好的分离度,实验采用梯度洗脱程序。为了延迟一些高极性化合物的保留时间,实验初期阶段的水相比例较高,而后逐渐降低水相的比例,使得化合物按极性由强到弱顺序,逐步被洗脱下来。当全部目标化合物洗脱出来以后,为了使一些强保留的弱极性杂质从柱子上洗脱下来,采用高有机相比例冲洗柱子。经过反复实验,综合考虑分离度和总分析时间,确定1.3中的梯度洗脱程序为最佳方案。图1所示为25种兽药在该条件下的总离子流图。
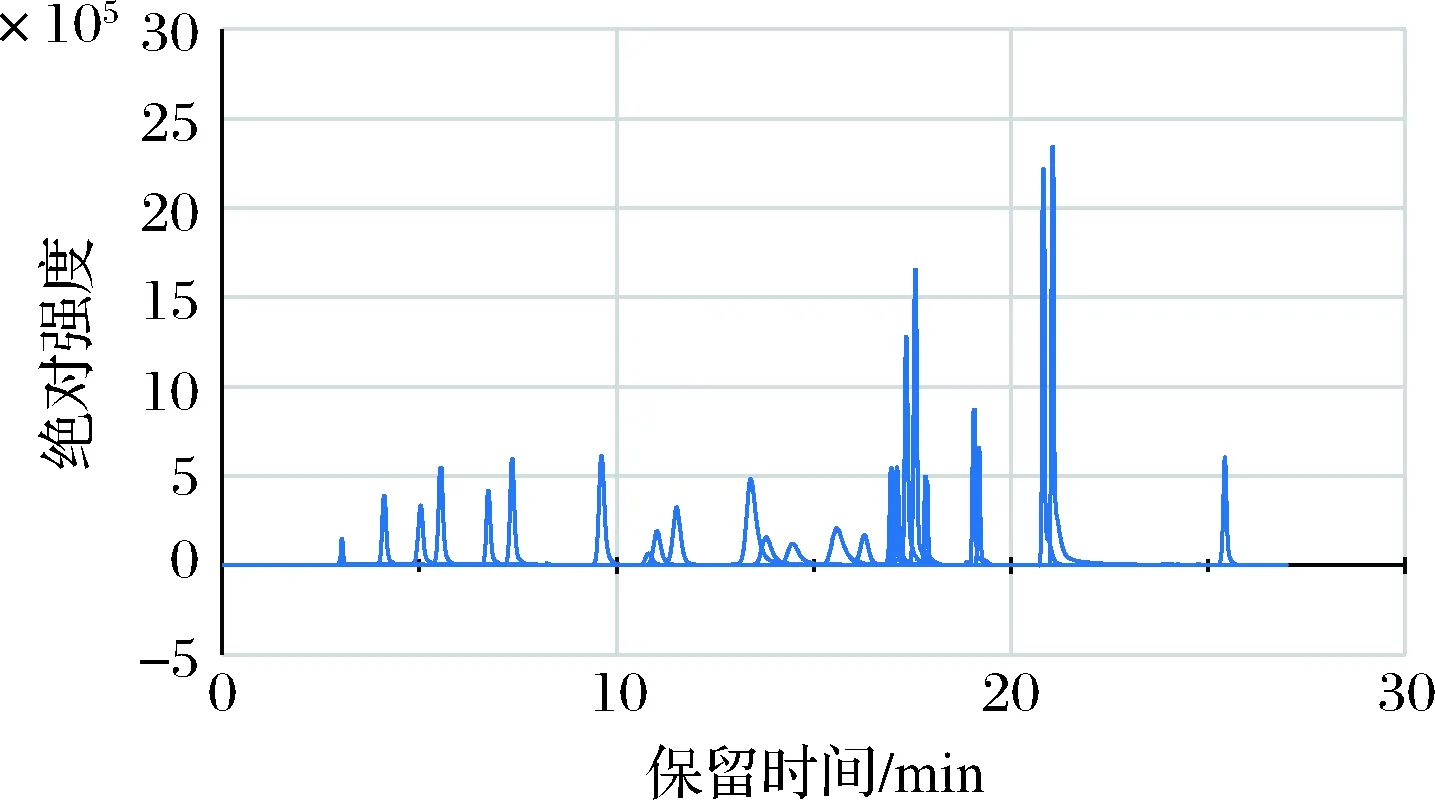
图1 25种兽药混合标准溶液的总离子流图(50 μg/L)Fig.1 Mass spectra of veterinary drug residues
2.1.2 质谱条件的确认
通过分别注射单一标准溶液,使用仪器自动优化的方法获得各个目标化合物的最优质谱条件。根据各个化合物的保留时间,分窗口采集信号,以便获得良好的灵敏度。
2.2 提取溶剂的选择
考虑到本文所涵盖的几类药物化学性质差异显著,本文比较了乙腈、甲醇、0.2%甲酸乙腈、0.1 mol/L Na2EDTA-Mcllvaine缓冲溶液+乙腈、0.1 mol/L Na2EDTA-Mcllvaine缓冲溶液+0.2%甲酸乙腈作为提取液时的提取效率。结果表明,0.1 mol/L Na2EDTA-Mcllvaine缓冲溶液+0.2%甲酸乙腈作为提取溶剂时几类化合物的综合提取效率最优。因此实验初步选定提取溶剂为:0.1 mol/L Na2EDTA-Mcllvaine缓冲溶液+0.2%甲酸乙腈。
实验又进一步比较了不同酸度的提取溶液对提取效率的影响,比较了0.2%、0.4%、1%、3%、5%、7%甲酸乙腈+0.1 mol/L Na2EDTA-Mcllvaine缓冲溶液作为提取溶剂时的实验效果,结果表明,在0.2%~7%,随着pH值的不断下降,喹诺酮类化合物的回收率逐步上升,但磺胺类化合物的回收率不断下降,其他几类化合物回收率先上升再下降。统筹考虑,最终选定0.1 mol/L Na2EDTA-Mcllvaine缓冲溶液+5%甲酸乙腈作为提取溶剂。
2.3 盐析剂和脱水剂的确定
实验比较了盐析剂NaCl和乙酸钠,脱水剂Na2SO4和MgSO4对实验结果的影响,并进一步确认了它们的用量。结果表明,加入乙酸钠和MgSO4会释放热量,造成一些兽药的回收率下降。实验最终确定NaCl作为盐析剂,用量为1 g;Na2SO4作为脱水剂,用量为4 g。
2.4 净化剂的确定
为了减少干扰物,降低基质效应的影响,需要对样品进行净化。常规的净化除杂填料有N-丙基乙二胺(primary secondary amine,PSA)、C18、—NH2吸附剂和石墨化炭黑(graphitizing of carbon black,GCB)等。PSA和—NH2主要用来除去脂肪酸、糖类等极性杂质;C18主要吸附非极性物质和油脂类杂质;GCB能吸附弱极性和具有苯环结构的化合物,会导致该类化合物回收率的下降,对本文所涉及的一些药物不适用。
本文比较了PSA、C18的除杂效果,结果表明:PSA能使磺胺类药物的回收率大幅降低,这可能是因为PSA吸附了该类化合物;C18能起到很好的净化作用,并有较好的回收率。因此,选用C18来去除杂质。
实验又进一步比较了C18和MgSO4不同用量配比时的除杂效果,分别为:30和200 mg,50和300 mg,100和600 mg,150和900 mg。结果表明50 mg C18和300 mg MgSO4时净化效果为满意,随着净化剂用量的进一步增加,净化效果没有明显提升。统筹考虑做样成本和除杂效果等原因,最终确定净化剂为50 mg C18和300 mg MgSO4。
2.5 方法学评价
2.5.1 线性关系和检出限
根据需要准确吸取一定量1.2中制备的标准储备液,配制成一系列质量浓度(0.1、0.2、0.5、1、5、10、50、100、200 μg/L)的混合标准工作液,在1.3的仪器条件下进行实验。以各个化合物的浓度和峰面积为横纵坐标,绘制线性关系曲线。结果得到25种化合物的相关系数范围0.990 2~0.999 9,说明25种兽药在0.1~200 μg/L具有良好的线性关系。
2.5.2 准确度和精密度
以草鱼样品为基质空白,进行5个重复3个水平(2、4和10 μg/kg)的添加回收实验,实验结果见表2。25种化合物的平均回收率为71.8%~107%,相对标准偏差(relative standard deviation,RSD)为4.4%~9.2%,该方法精密度和准确度能满足现有法规的要求,可用于准确定量,适用于实际样品的测定。
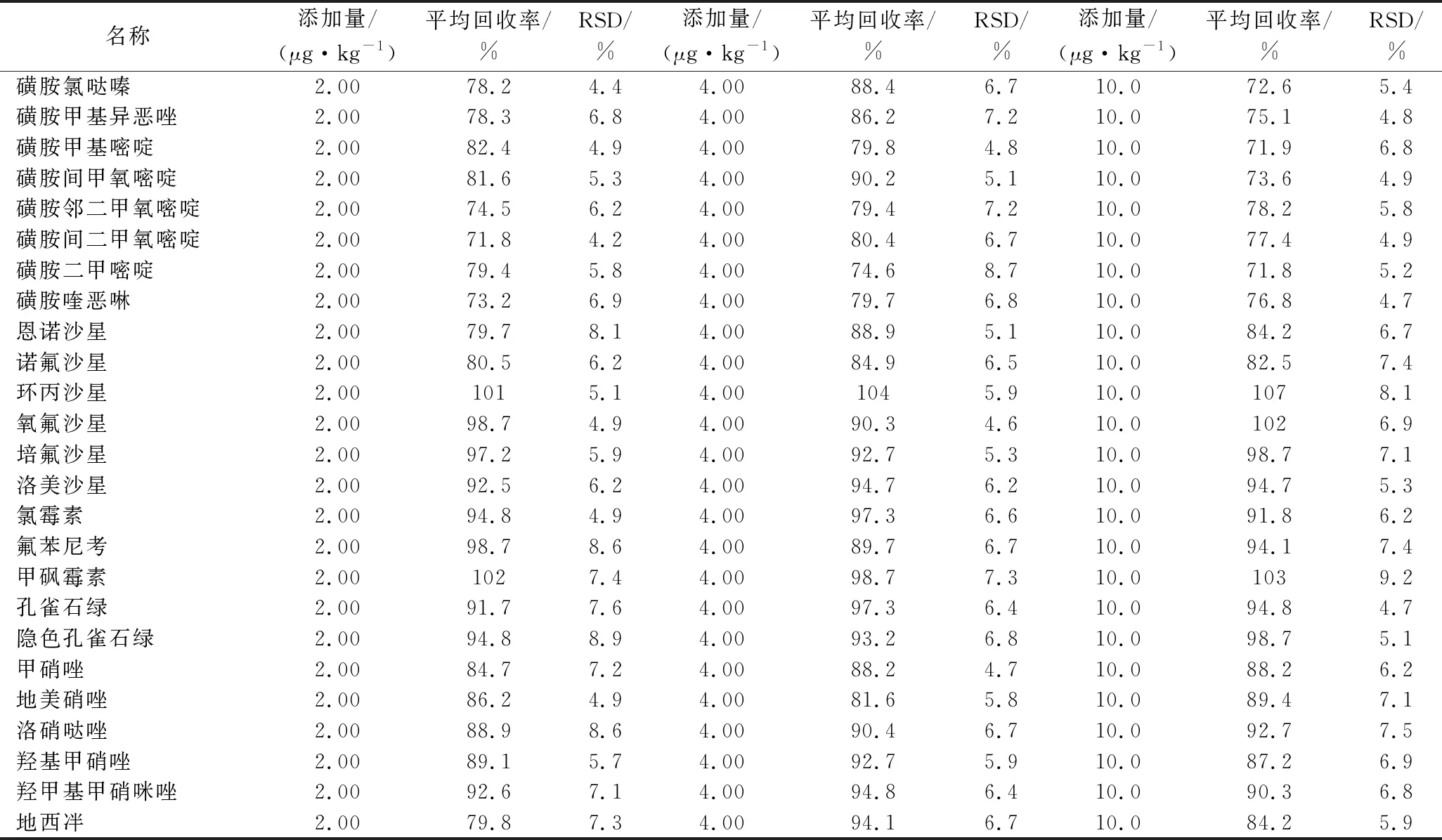
表2 兽药残留添加回收表Table 2 Recoveries and precisions of veterinary drug residues
2.6 实际样品的测定
本课题组在2019年的食品安全监督抽查工作中,鱼类产品共检出恩诺沙星、氧氟沙星、磺胺嘧啶若干份,其中恩诺沙星最低值16 μg/kg,最高值596 μg/kg,氧氟沙星最低值28.9 μg/kg,最髙值476 μg/kg,磺胺类总量(磺胺嘧啶、磺胺二甲嘧啶、磺胺甲基嘧啶、磺胺甲恶唑、磺胺间二甲氧嘧啶、磺胺邻二甲氧嘧啶、磺胺间甲氧嘧啶、磺胺氯哒嗪、磺胺喹恶啉之和)最低值13.2 μg/kg,最髙值113 μg/kg。使用本文所述的方法,对检出的阳性样品与国标法进行方法比对试验,实验结果采取F检验方法进行单因素方差分析,结果显示2种方法在显著性水平为0.05水平时,无显著性差异(表3)。

表3 国标法与本文所建立方法检测结果对比表 单位:mg/kg
3 结论
本研究建立了使用QuEChERS前处理方法,同时测定鱼肉中磺胺类、喹诺酮类、酰胺醇类等药物多残留的检测技术。该技术的前处理方法简单、快速,方法学评价结果符合兽药残留的相关要求。使用本文建立的方法对实际工作中检出的阳性样品进行测定,结果表明该方法和国标法测定结果无显著差异,该方法可以用于实际样品的检测。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
核化学与放射化学(2022年2期)2022-04-28
中国饲料(2022年5期)2022-04-26
海洋通报(2020年5期)2021-01-14
湖南农业(2020年7期)2020-09-16
国外畜牧学·猪与禽(2019年8期)2019-11-11
智富时代(2019年7期)2019-08-16
智富时代(2019年7期)2019-08-16
肥料与健康(2018年6期)2018-03-04
云南农业(2015年10期)2015-03-19
