
氧化亚油酸对肌原纤维蛋白胶凝行为及热诱导凝胶体外消化率的影响
2021-10-22 00:21李保玲李颖朱振宝黄峻榕曹云刚
食品与发酵工业 2021年19期
李保玲,李颖,朱振宝,黄峻榕,曹云刚
(陕西科技大学 食品与生物工程学院,陕西 西安,710021)
肉及其制品的质地特征是影响消费者做出购买决定的重要因素,占肌肉蛋白总量55%~65%的肌原纤维蛋白因为其优异的凝胶和乳化能力,对最终加工肉制品的质地特性起着主要决定作用[1]。因此在肉制品加工过程中,蛋白结构特征的变化及凝胶精细网络的形成受到了广泛关注[2-3]。在肉类食品加工过程中,肌肉细胞结构的破坏、分子氧的渗透以及游离铁和血红素铁的释放不可避免地导致了脂肪和蛋白质的氧化。长期以来人们对脂质的氧化进行了深入的研究,然而蛋白质作为活性氧(reactive oxygen species,ROS)或者其他过氧化物作用的靶点却一直被忽视[4-5]。之前的研究主要聚焦于蛋白质氧化在年龄相关疾病中的作用[6-7],但目前越来越多的研究开始探讨蛋白质氧化对食品质量的影响。ROS 被认为是与氨基酸侧链基团的氧化修饰有关的原位因素,通常会导致蛋白质结构与功能性质的改变,包括持水性能、凝胶性能以及乳化性能等[5,8]。
由于原料肉中蛋白质和不饱和脂肪大量共存以及加工行为的破坏性作用,脂肪和蛋白的氧化极可能以相关联的方式发生。相关研究表明,脂质氢过氧化物、脂质自由基以及衍生的醛类物质等都可以促进蛋白氧化的发生[9-10]。关于脂质氧化引起的肌原纤维蛋白(myofibrillar protein,MP)生化指标和蛋白消化率的变化已有大量研究报道[11-14],但是由脂质氧化产物导致的MP凝胶性能改变的现有研究结果并不一致,同时前人研究多集中于氧化对生蛋白消化率的影响,而现实生活中人们对肉食的摄入主要以熟食为主。因此,本实验主要研究蛋白暴露在脂肪氧合酶——亚油酸氧化体系中所引起的MP结构以及凝胶性能的变化和氧化对蛋白热诱导凝胶体外消化率的影响,以此进一步了解肉类及其制品中脂质氧化和蛋白质氧化之间的关系。
1 材料与方法
1.1 实验材料
猪外脊肉(Longissimuslumborum),当地超市(陕西西安);大豆脂肪氧合酶、胰蛋白酶,美国Sigma-Aldrich公司;亚油酸(>95%)、胃蛋白酶,上海阿拉丁化学有限公司;其他试剂至少为分析纯。
1.2 仪器与设备
HR/T20MM立式高速冷冻离心机,湖南赫西仪器装备有限公司;TA.Plus物性测试仪,英国Stable Micro System公司;Fluoro Max-4 荧光分光光度计,日本 Horiba 公司;Mastersizer 2000激光粒度分析仪,英国Malvern Instruments有限公司;CM-5分光测色计,柯尼卡美能达(中国)投资有限公司;Haake-Mars 60 流变仪、DXR2显微共聚焦拉曼光谱仪,德国Thermo Fisher Scientific公司;FEI Q45+EDAX Octane Prime环境扫描电子显微镜,美国FEI公司;安东帕LitesizerTM500纳米粒度及Zeta电位分析仪,奥地利Anton Paar 公司。
1.3 MP的提取及样品制备
1.3.1 MP的提取
肌原纤维蛋白的提取参照PARK等[13]的方法,使用双缩脲法以牛血清白蛋白(bovine serum albumin,BSA)作为标准测定蛋白浓度,将所得MP置于碎冰浴4 ℃保存并在48 h内使用。
1.3.2 蛋白的氧化处理
用25 mmol/L的磷酸盐缓冲液(含0.4 mol/L NaCl,pH 6.2)将MP稀释至45 mg/mL,将MP溶液分散于脂肪氧化体系(含0.5~10.0 mmol/L 亚油酸,3 750 U/mL脂肪氧合酶,终浓度)使MP终浓度达到30 mg/mL,并于4 ℃反应12 h。通过添加Trolox(终浓度 1 mmol/L)来终止氧化反应,未添加氧化体系的MP溶液为空白对照。
1.4 脂质氧化的测定
参照SALIH等[15]的方法测定2-硫代巴比妥酸反应物(thiobarbituric acid reactive substances,TBARS)含量,并以此来反映MP悬液中的脂质氧化程度。
1.5 MP的化学及结构变化
1.5.1 氨基酸残基侧链修饰
羰基含量采用CAO等[16]描述的2,4-二硝基苯肼(2,4-dinitrophenylhydrazine,DNPH)比色法进行测定。
总巯基含量按照ELLMAN[17]的方法,采用5,5’-二硫代双(2-硝基苯甲酸)[5,5'-dithiobis-(2-nitrobenzoic acid),DTNB]进行测定。
自由氨基含量参照HABEEB[18]的方法,通过2,4,6-三硝基苯磺酸(2,4,6-trinitrobenzenesulfonic acid,TNBS)试剂进行测定,同时使用一系列浓度的L-亮氨酸在相同条件下制成标准曲线,并通过绘制的标准曲线来计算蛋白样品的自由氨基含量。
1.5.2 蛋白构象变化
内源性色氨酸荧光的测定:使用25 mmol/L的磷酸盐缓冲液(含0.4 mol/L NaCl,pH 6.2)将样品稀释至0.4 mg/mL,利用荧光分光光度计于波长283 nm激发并记录290~400 nm的发射光谱,激发和发射狭缝宽度均设置为1.5 nm。相同条件下记录溶剂发射光谱,并从样品发射光谱中扣除以排除干扰。
1.6 Zeta电位和粒度测定
Zeta电位:利用电位分析仪进行测定,使用25 mmol/L磷酸盐缓冲液(含0.4 mol/L NaCl,pH 6.2)将样品稀释至1 mg/mL,取1 mL放入Univette样品池,平衡时间设定为1 min,设置溶剂(磷酸盐缓冲液)折射率为1.331 8,电泳温度维持在25 ℃,梯度重复3次,取其平均值。
粒度测定:使用激光粒度分析仪在25 ℃下采用静态光散射分析MP样品的粒径分布,去除测量背景后将稀释后的MP样品(2 mg/mL)分散在蒸馏水(分散介质)中,直至遮光效果达到10%~13%,设置MP颗粒折射率为1.434,分散剂折射率为1.330,测定样品的平均粒度(d3,2和d4,3)。
1.7 溶解度
使用25 mmol/L的磷酸盐缓冲液(含0.4 mol/L NaCl,pH 6.2)将肌原纤维蛋白样品稀释至 2 mg/mL,然后在5 000×g条件下离心(15 min,4 ℃)。取上清液根据CAO等[1]的方法测定得到的蛋白样品的溶解度。
1.8 十二烷基硫酸钠聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,SDS-PAGE)
采用SDS-PAGE分析了蛋白质组分的氧化交联、聚合或降解情况。实验参照CAO等[19]的方法进行,使用4%浓缩胶和12%分离胶在还原(+DTT)和非还原(-DTT)条件下进行凝胶电泳,每孔上样量20 μg,染色脱色后拍照并对电泳条带进行分析。
1.9 凝胶性能测定
1.9.1 动态流变性能测定
将不同处理的MP样品离心脱气后(30 mg/mL,1 000×g,1 min),取适量样品均匀涂布于流变仪的平板上,选用平行板传感器P35(直径35 mm)下压至1 mm处,用纸去除周围多余样品,在边缘涂上硅油防止水分蒸发,在振荡模式下从 20 ℃加热到 75 ℃,升温速率:1 ℃/min,振荡频率:0.1 Hz,控制最大应变:0.02。凝胶性能通过储存模量(G′)来进行评价。
1.9.2 蒸煮损失、凝胶强度和白度
将MP样品(5 g,30 mg/mL)置于小玻璃瓶中使用保鲜膜密封后置于水浴锅中,以1 ℃/min的升温速率从20 ℃加热至75 ℃,并在75 ℃保温10 min,取出后立刻置于冰水混合物中冷却30 min,随后放入4 ℃冰箱冷藏过夜。蒸煮损失、凝胶强度和白度的测定参照CAO等[1]的方法进行。
1.9.3 MP凝胶微观结构
将制备的MP凝胶切成小块(5 mm×5 mm×5 mm),用含2.5%(体积分数)戊二醛的磷酸盐缓冲液(pH 7.4)固定4 h,使用磷酸盐缓冲液清洗3次,然后通过一系列浓度的乙醇溶液(50%,70%,90%,95%,100%体积分数)进行梯度脱水,每次30 min。在-70 ℃对样品进行冷冻干燥,将干燥的样品喷金后使用环境扫描电子显微镜(environmental scanning electron microscope,ESEM)观察其微观结构,加速电压15 kV,放大倍数3 500倍。
1.9.4 MP凝胶的疏水相互作用和氢键作用
采用拉曼光谱对MP凝胶中760 cm-1附近的拉曼谱带归一化强度和830、850 cm-1处酪氨酸双谱带的相对强度进行了表征[20],探究氧化对MP凝胶疏水相互作用和氢键作用的影响,光谱数据由OMNIC软件收集后使用LabSpec对所得光谱进行平滑、基线校正并用在1 003 cm-1处的苯丙氨酸环带进行归一化处理[21]。
1.9.5 MP凝胶的体外消化
参照曹云刚[22]的方法模拟胃-肠道消化对MP凝胶进行体外消化评价。取冷却过夜的MP凝胶及其流出液5 g,磨碎后加入适量10 mmol/L的HCl溶液,在11 000 r/min条件下均质30 s,添加猪胃蛋白酶溶液(1 mg/mL,溶于10 mmol/L HCl溶液),所得混合溶液蛋白质量浓度为 3 mg/mL,胃蛋白酶浓度4%(质量分数,以蛋白含量为基准计算),pH 2.0。混匀后于37 ℃水浴锅中酶解1 h,而后使用1 mol/L的NaOH溶液调节pH值至7.5来结束反应,而后添加4%(质量分数,以蛋白含量为基准计算)的胰酶溶液,继续在37 ℃水浴锅中反应2 h,反应结束后于沸水浴中加热5 min以达到灭活的目的。消化反应期间分别在30、60、90、120、180 min时取样,加入同等体积的30% (质量分数)三氯乙酸来终止反应沉淀蛋白,混合液在4 ℃冰箱中过夜,以11 000×g离心10 min后弃去上清液,于沉淀中加入1 mol/L的NaOH溶液来复溶蛋白沉淀,随后使用双缩脲法测定蛋白浓度。体外消化率计算如公式(1)所示:
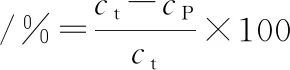
(1)
式中:ct和cp分别指总蛋白浓度以及体外消化后三氯乙酸沉淀的蛋白浓度。
1.10 数据分析
所有的实验至少独立重复2次,每批次至少有3个独立样品。实验数据使用Statistix 9.0分析软件进行方差分析,采用LSD对多组数据进行显著性分析,P<0.05表示差异有统计学意义。数据以平均数±标准差(mean±SD)表示,并使用Sigma Plot 12.5软件进行绘图。
2 结果与分析
2.1 脂质氧化
相关研究发现,多不饱和脂肪酸(亚油酸、亚麻酸等)在脂肪氧合酶的催化下发生脂质过氧化反应,形成自由基中间体和相关脂质氧化产物,进而促进MP氧化[12]。本研究采用TBARS法通过测定脂质氧化二级产物的含量来评价脂质氧化程度。如表1所示,未氧化MP的TBARS值为0.94 mg/kg蛋白,这与先前研究报告的值(0.79 mg/kg)相似[23]。总的来说,随着OLA浓度的增加,脂质氧化MP样品TBARS值不断增加,在OLA浓度为10.0 mmol/L时,TBARS值达到最大(4.86 mg/kg),是对照样品的5倍。这一结果说明OLA浓度的增加促进了脂质氧化反应的发生。
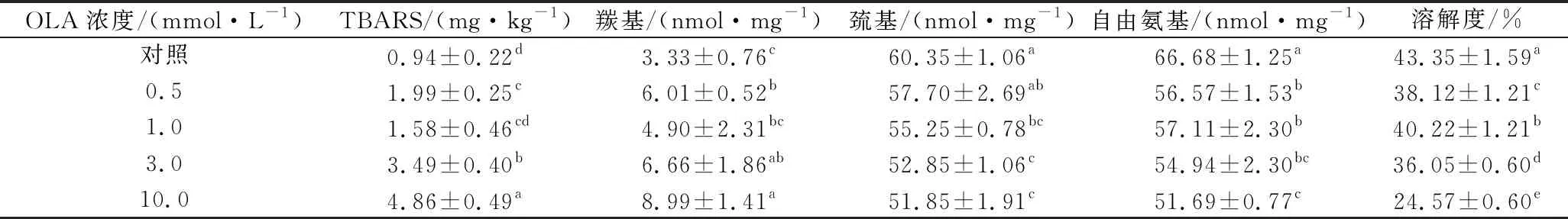
表1 不同OLA浓度下肌原纤维蛋白理化性质的变化Table 1 Changes in physicochemical properties of myofibrillar protein at different concentrations of OLA
2.2 MP样品氨基酸残基的侧链修饰情况
2.2.1 羰基
蛋白质羰基含量的变化是反映蛋白质氧化程度的重要指标,它主要由氨基酸侧链残基和肽键的氧化断裂产生[5]。OLA浓度对MP羰基含量的影响如表1所示,未氧化MP的羰基含量为3.33 nmol/mg,这一结果与JONGBERG等[24]研究的未氧化猪肉MP的羰基值相接近。然而,也有部分研究报道的未氧化猪肉MP的羰基值较低[11,14],推测实验数据的不一致与实验所用猪肉的品种及屠宰方式有关。与TBARS含量变化趋势一致,随着OLA浓度的增加,脂质氧化MP中羰基也在不断生成,在OLA浓度为10.0 mmol/L时羰基含量达到最大值(8.99 nmol/mg,P<0.05),其他研究者也报道了脂质氧化MP中羰基含量的类似变化趋势[11,14]。这些结果表明脂质氧化促进了蛋白氧化的发生,同时也证实了脂质氧化产物可以有效催化蛋白氧化的观点。
2.2.2 总巯基
MP中巯基含量丰富,其暴露在氧化环境中易被转变成分子内或分子间二硫键及其他相关化合物,使MP中总巯基含量下降,进而对蛋白的结构和功能性质造成影响[25]。如表1所示,未氧化MP中的总巯基含量为60.35 nmol/mg,与先前研究报道的值相似[14,19]。当MP暴露于脂质氧化体系中,随着OLA浓度的增加,蛋白的总巯基含量稳步下降,与未氧化MP相比,OLA浓度为10.0 mmol/L时,总巯基含量下降约14.1%。类似的氧化引起MP总巯基含量降低的研究在各种氧化体系中已被广泛报道[26-27]。
2.2.3 自由氨基
赖氨酸残基的ε-NH2基团在氧化过程中通过脱氨作用转化为羰基,然后形成的羰基与氨基反应生成席夫碱,进而导致游离氨基的含量持续下降[28]。如表1所示,MP暴露于脂肪氧化体系后游离氨含量显著下降(P<0.05)。与未氧化MP相比,经过不同浓度OLA处理后(0.5、1.0、3.0和10.0 mmol/L)MP样品自由氨基含量分别减少了15.2%、14.4%、17.6%和22.5%。脂质氧化导致了MP中羰基的形成、巯基和游离氨基的损失,表明脂质氧化产生的ROS对蛋白质具有广泛的氧化攻击作用。
2.3 MP内源性色氨酸荧光强度的变化
由于色氨酸残基对周围微环境的敏感性,内源性色氨酸荧光特性通常用于反映蛋白质结构的变化[29]。当蛋白质处于折叠状态时,MP具有较大的荧光强度和较短的最大发射波长;当蛋白质结构展开时,色氨酸残基暴露于极性环境中,其荧光强度降低、最大发射波长红移(变长)。如图1所示,随着OLA浓度的增加,MP最大发射波长处的荧光强度逐渐降低,特别是当OLA浓度超过1.0 mmol/L时(P<0.05)。与此同时,与未氧化MP相比,当OLA浓度增加到10.0 mmol/L时,MP的最大发射波长从328.5 nm变为329.6 nm。推测可能的原因是随着氧化的进行,蛋白质结构逐渐展开,MP中的色氨酸残基等荧光基团暴露在极性环境中,导致MP样品的色氨酸荧光强度降低。
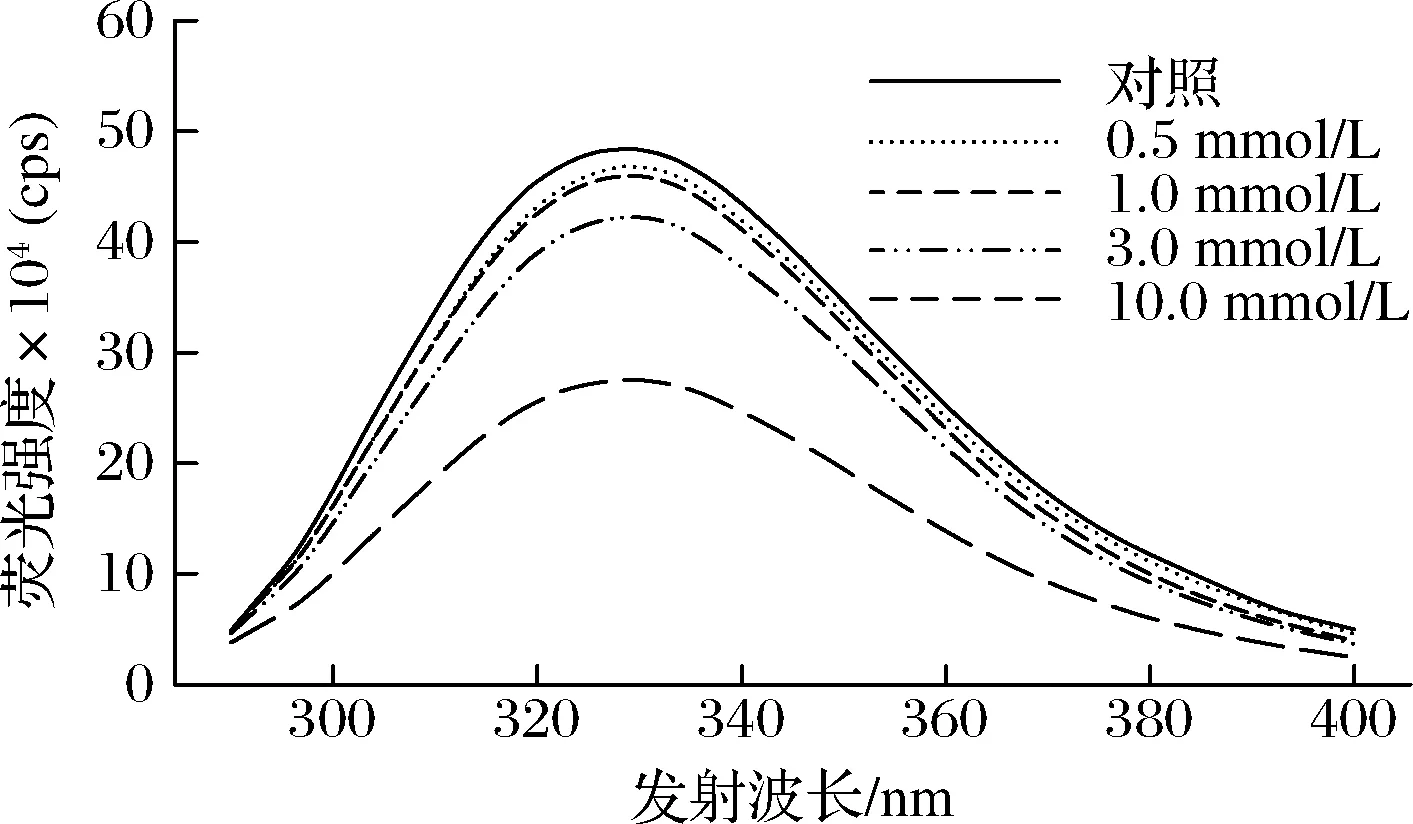
图1 不同OLA浓度下MP的色氨酸荧光强度变化Fig.1 Changes in tryptophan fluorescence intensity of myofibrillar protein at different concentrations of OLA
2.4 MP样品Zeta电位和粒径的变化
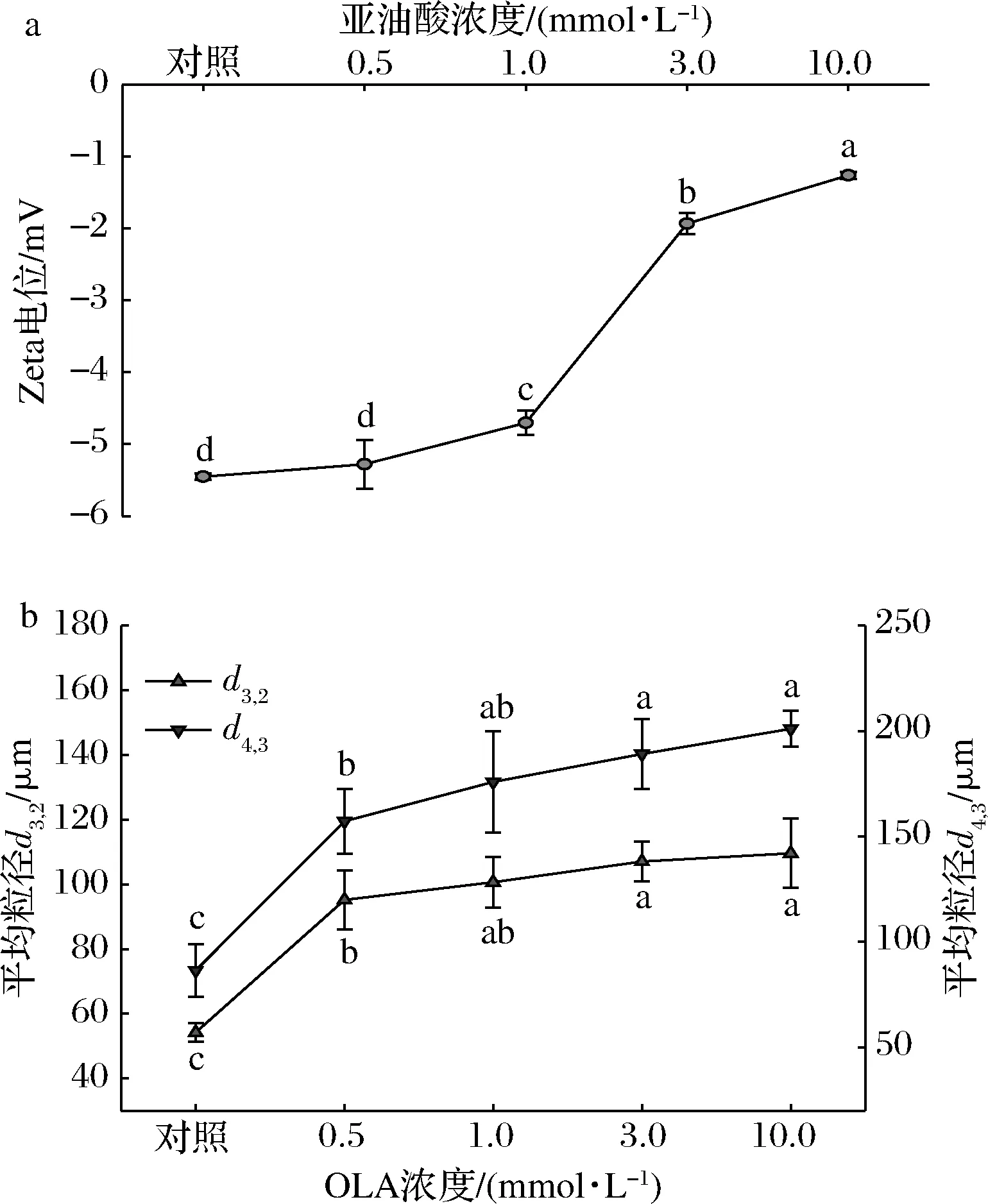
Zeta电位反映了MP溶液中蛋白粒子间的静电相互作用,它与MP溶液的稳定性密切相关。本研究中MP样品的Zeta电位均为负值(图2-a),这与MP溶液的pH值高于蛋白等电点有关,阴离子主要来自于蛋白质颗粒表面的酸性氨基酸残基(如天冬氨酸、谷氨酸等)。
a-Zeta电位;b-粒径图2 不同OLA浓度下MP的Zeta电位和粒径变化Fig.2 Changes in Zeta potential and mean particle diameter of myofibrillar protein at different concentrations of OLA
与未氧化MP相比,随着OLA浓度的增加,MP的Zeta电位绝对值不断减小,说明蛋白粒子间的静电作用力随着氧化程度的增强而不断减弱。
如图2-b所示,通过测定MP样品的平均粒径d3,2和d4,3来评估氧化诱导的蛋白聚集行为。从图中可以看到,随着OLA浓度的增大,MP样品的平均粒径不断增大。结合前文MP样品溶液的内源性色氨酸荧光强度以及Zeta电位结果发现:氧化导致蛋白质去折叠化,暴露出带电荷的氨基酸残基与氧化产物反应生成电中性物质,从而使蛋白粒子间静电相互作用减弱、蛋白质分子间的交联聚集加剧,引起蛋白平均粒径的增加。这一结果与周非白[30]关于羟自由基氧化体系诱导的MP氧化以及BAO等[31]使用不同浓度次氯酸诱导蛋白氧化实验得到的MP平均粒径变化趋势相似。
2.5 MP溶解度的变化
MP的溶解度反映了蛋白质交联、聚集程度的变化。脂质氧化程度对MP溶解度的影响如表1所示,未经氧化处理的MP溶解度为43.35%,而当MP暴露于脂质氧化体系后蛋白溶解度持续降低(P<0.05)。与未氧化MP相比,在OLA浓度逐步增加至0.5、1.0、3.0、10.0 mmol/L时,MP的溶解度分别下降了12.1%、7.2%、16.8%和43.3%。推测氧化后MP溶解度的降低主要与以下因素有关:疏水基团的暴露(图1)、二硫键和其他共价键的形成,这些变化导致蛋白质之间的相互作用增强,分子间的交联聚集加剧(图2-b),从而降低了蛋白样品的溶解度。
2.6 SDS-PAGE
不同浓度OLA处理下MP的SDS-PAGE图谱如图3所示。在非还原条件下(图3-a,-DTT),随着OLA浓度的增加,肌球蛋白重链(myosin heavy chain,MHC)和肌动蛋白(actin)条带强度稳步下降,同时在浓缩胶的顶部观察到高分子质量聚合物的聚集,这一结果进一步解释了亚油酸诱导的脂质氧化导致蛋白质溶解度降低的原因。如图3-b(+DTT)所示,在还原条件下堆积在浓缩胶顶端的聚合物几乎完全消失,MHC和actin条带强度明显增强,这一结果意味着氧化诱导形成的聚合物主要与MHC和肌动蛋白有关,同时也表明脂质氧化引起的MP总巯基含量的下降主要是由于二硫键的形成[26,32]。
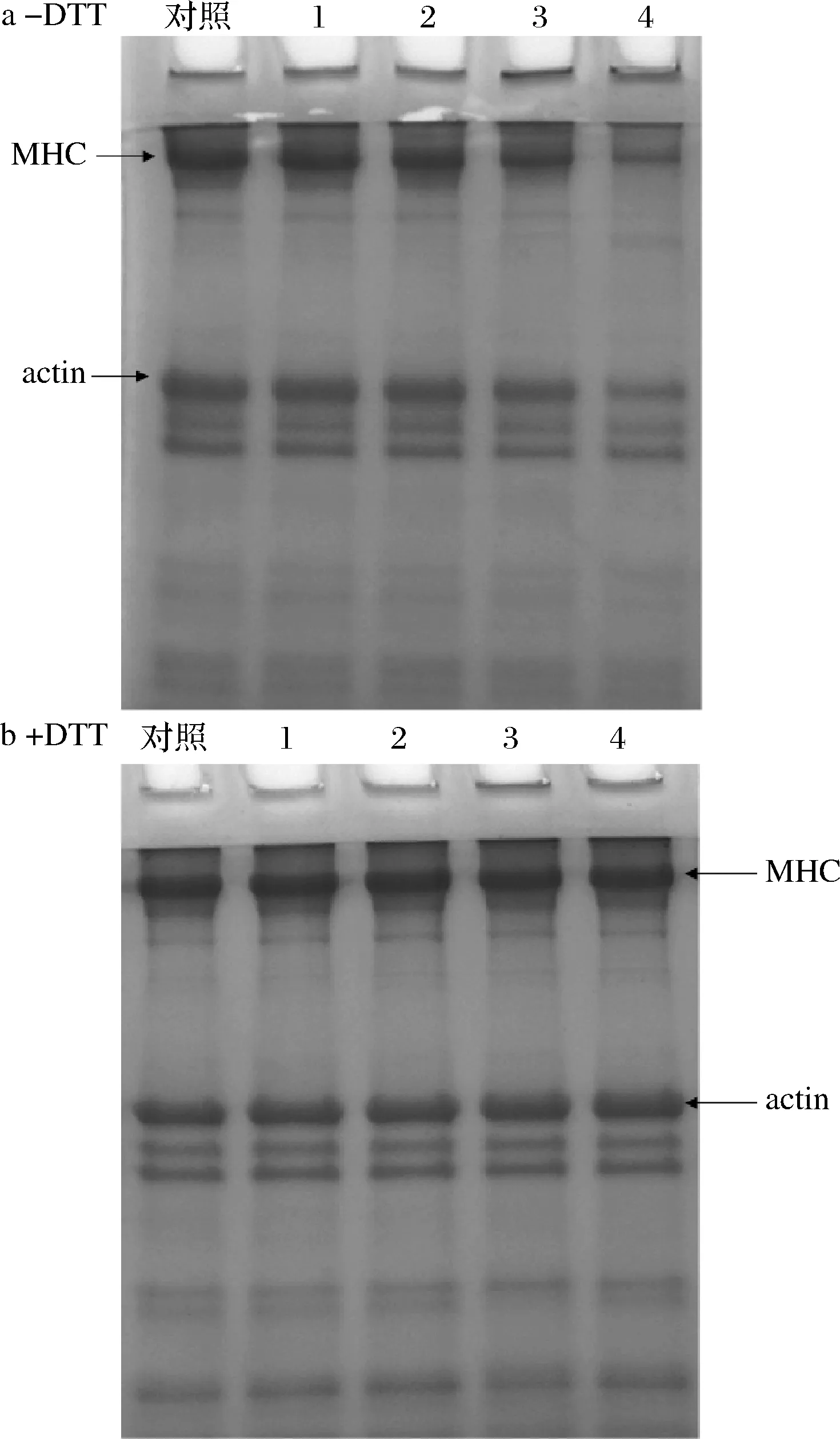
1-0.5 mmol/L;2-1.0 mmol/;3-3.0 mmol/L;4-10.0 mmol/L图3 不同OLA浓度下MP SDS-PAGE图谱的变化Fig.3 Changes of SDS-PAGE patterns of myofibrillar protein at different concentrations of OLA
2.7 MP凝胶性能的变化
2.7.1 动态流变行为
MP悬浮液的流变特性可以反映蛋白质氧化对凝胶形成过程以及凝胶弹性的影响。不同氧化处理的MP悬浮液在加热过程中的流变规律如图4所示。未氧化MP样品呈典型的G′曲线,在49 ℃左右达到一个过渡峰,在53 ℃左右出现一个低谷,随后在53~75 ℃稳定上升。相关报道表明G′在49 ℃左右的第一个转变峰是由肌球蛋白头部的变性和聚集引起的[33],而约53 ℃出现的波谷则与轻酶解肌球蛋白变性有关,此后G′的持续增加则归因于永久性和不可逆肌球蛋白丝或复合物的形成[33-34]。OLA的存在显著降低了初始的G′和过渡峰G′值的大小,然而最终G′值并没有显著变化。
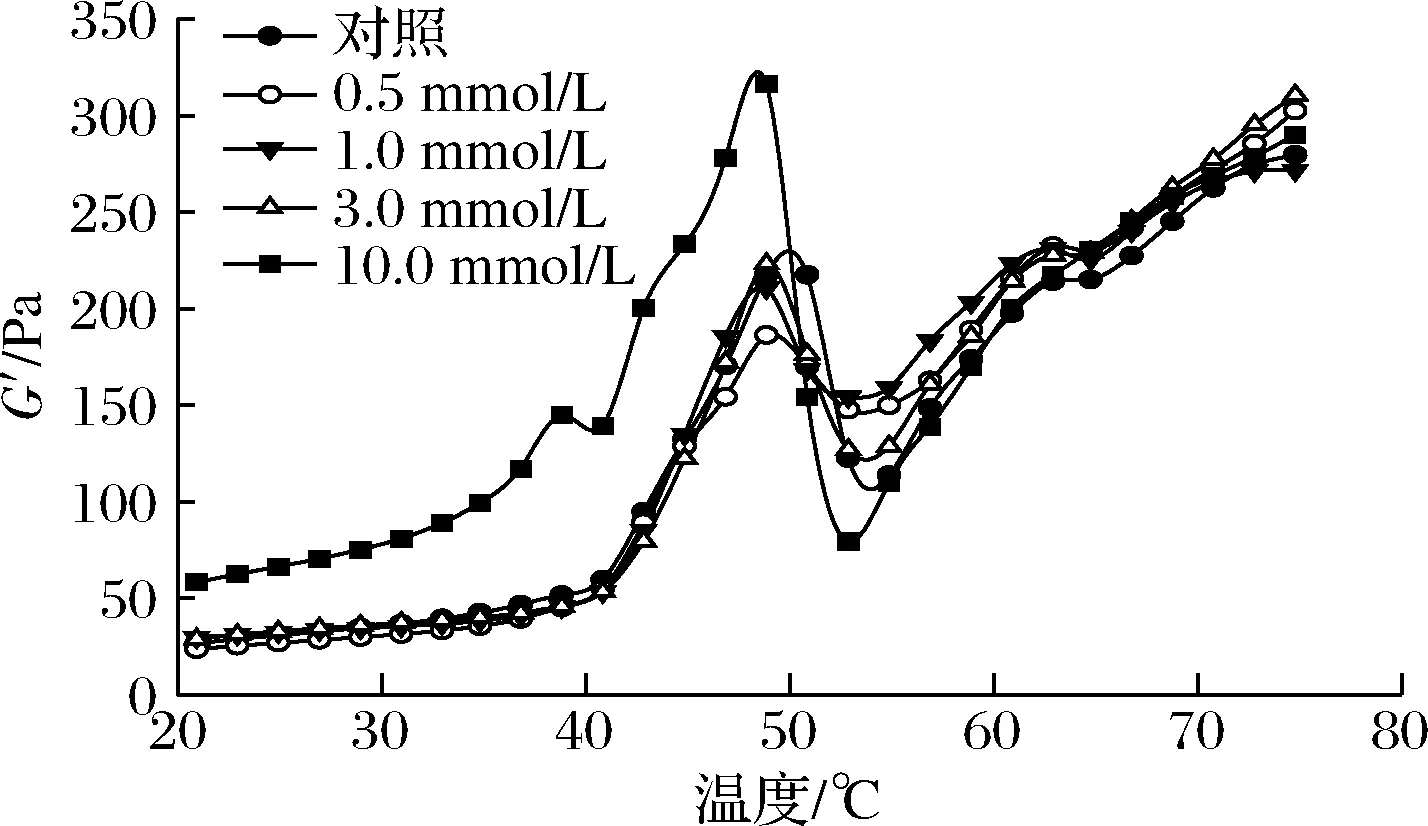
图4 不同OLA浓度下MP的流变性能Fig.4 Rheological properties of myofibrillar protein at different concentrations of OLA
亚油酸诱导的脂质氧化导致蛋白官能团的修饰、蛋白质构象的变化以及MP聚集等现象的发生,从而改变了MP在加热过程中的动态流变行为。
2.7.2 蒸煮损失、凝胶强度和白度
如表2所示,随着OLA浓度的增加,MP凝胶的蒸煮损失逐渐上升。与未氧化MP相比,在向MP样品分别添加0.5、1.0、3.0和10.0 mmol/L的OLA后,MP凝胶的蒸煮损失分别增加46.6%、73.8%、78.6%和131.1%(P<0.05)。与蒸煮损失的变化趋势相反,MP的凝胶强度随着OLA浓度的增加而逐渐降低,经0.5、1.0、3.0和10.0 mmol/L的OLA处理后,MP凝胶的初始断裂力分别为对照组的82.1%、76.2%、75.0%和73.8%。推测随着氧化强度的增加,MP分子间的交联聚集加剧、溶解度不断下降,这种情况影响了MP均匀牢固的凝胶网络结构的形成,造成了MP蒸煮损失的增加以及凝胶强度的降低[19,35]。UTRERA等[36]研究发现氧化导致蛋白质羰基化,从而改变蛋白质的电荷特性,导致肌肉蛋白质的溶解度、保水性和凝胶特性降低,与本研究结果一致。
已有文献报道表明蛋白的凝胶白度与蛋白质的变性程度相关[37]。如表2所示,凝胶白度随OLA浓度的增加而显著降低,经过0.5、1.0、3.0和10.0 mmol/L的OLA处理后的样品凝胶白度分别为对照组的98.8%、98.4%、97.0%和95.5%。
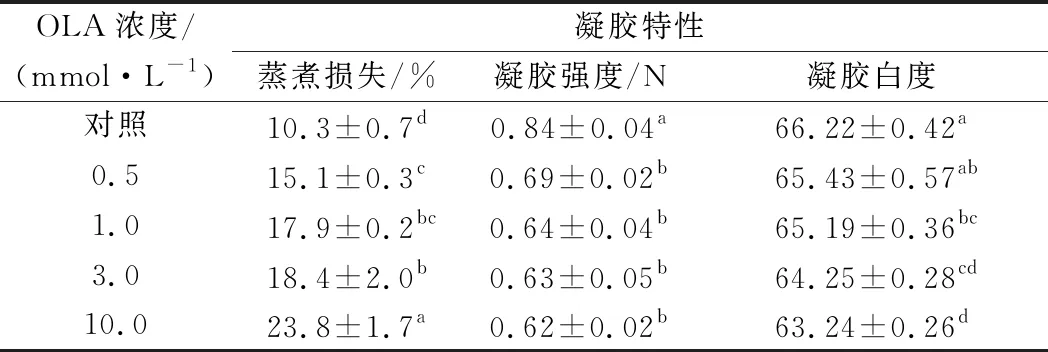
表2 不同OLA浓度下MP的凝胶性能变化Table 2 Changes in gelling properties of myofibrillar protein at different concentrations of OLA
周非白[30]研究发现随着过氧自由基含量的增加,MP的凝胶白度显著降低,与本研究结果一致。XIA等[38]发现猪肉MP的凝胶白度随肌肉冷冻-解冻循环次数的增加而显著降低,作者认为这一现象与脂质氧化有关。而在真实的肉类体系中,ESTÉVEZ等[39]发现法兰克福香肠的颜色与肌肉蛋白的氧化直接相关。因此,本研究中MP凝胶白度的降低应与脂质氧化和蛋白氧化相关。
2.7.3 MP凝胶的微观结构
蛋白质凝胶微观结构的表征是研究MP凝胶特性的重要手段。不同OLA浓度对MP热诱导凝胶微观结构的影响如图5所示,未氧化MP形成的凝胶网络结构致密,微孔分布均匀。而OLA的添加对MP凝胶微观结构的影响主要与其浓度相关,低浓度OLA的添加(0.5~1.0 mmol/L)对MP凝胶的微观结构没有显著影响,而当OLA浓度从3.0 mmol/L增加到10.0 mmol/L时,MP凝胶的微观结构发生显著变化。当OLA浓度达到10.0 mmol/L时,MP凝胶的网络结构变得粗糙且不规则,孔隙变大,凝胶网络结构受到严重破坏。而MP凝胶松散、不规则的结构也导致了MP凝胶持水性能和凝胶强度的降低。加热前MP的高强度氧化引起的蛋白过度聚集和溶解度降低是导致MP凝胶性能下降以及微观结构粗糙不规则的原因。

图5 不同OLA浓度下MP凝胶的微观结构图Fig.5 Microstructure of myofibrillar protein gels at different concentrations of OLA
2.7.4 拉曼光谱分析
760 cm-1(I760)附近的拉曼谱带归一化强度变化归属于色氨酸残基的伸缩振动,其对色氨酸残基周围的微环境变化极其敏感,因此可用于研究MP凝胶的疏水相互作用[40]。如表3所示,与未氧化MP凝胶相比,经过0.5 mmol/L的OLA处理后MP凝胶的I760值增加了约41.8%(P<0.05),这一结果表明轻度氧化增强了MP凝胶的疏水相互作用,然而随着OLA浓度的进一步增加,MP凝胶的I760值逐渐降低,表明过度氧化减弱了MP凝胶的疏水相互作用。
830和850 cm-1酪氨酸双谱带的相对强度(I850/I830)变化反应了苯环上酚羟基(—OH)的状态和氢键的性质[40]。当双峰的相对强度(I850/I830)比值较高(0.9~2.5),表明酪氨酸残基暴露在水溶液或其他极性微环境中或者同时作为氢键受体以及中弱等氢键供体进行传导,而较低的相对比值则表明酪氨酸残基埋藏在疏水环境中或仅作为氢键供体来增强氢键作用[21]。如表3所示,I850/I830的比值在0.982~1.036,这说明酪氨酸残基主要暴露在水溶液中并同时参与了中、弱氢键作用。需要注意的是,随着OLA浓度的增加,I850/I830的比值由0.982增加到1.036,表明随着氧化程度的加深,酪氨酸残基的苯环上更多的—OH暴露于水环境中并与水分子形成氢键[41]。换言之,MP凝胶中蛋白-蛋白氢键随着氧化程度的增加而减少。

表3 不同OLA浓度下MP凝胶I760的归一化强度和I850/I830比率Table 3 Normalized intensity of the 760 cm-1band and ratio of 850/830 cm-1 doublet bands of myofibrillar protein gels at different concentrations of OLA
2.7.5 MP凝胶的体外消化测定
使用模拟胃和肠道消化的消化酶(胃蛋白酶、胰酶)来测定不同OLA浓度下MP热诱导凝胶的消化率,结果如表4所示。在胃蛋白酶消化阶段,与未氧化MP热诱导凝胶相比,凝胶制备前的氧化处理对其最终消化率并没有显著性影响,当OLA浓度过大时(3.0~10.0 mmol/L),MP凝胶的消化率有少许下降。在胰蛋白酶消化阶段,与未氧化MP热诱导凝胶相比,OLA浓度为0.5 mmol/L时,蛋白在180 min时的消化率无显著变化(P>0.05),而当OLA浓度>0.5 mmol/L时,MP凝胶的消化率显著降低。这一结果也与周非白[30]在研究氧化对MP体外胃肠道消化率的结果类似,作者认为过度氧化条件下蛋白形成聚集体,同时造成了氨基酸的损失,因此导致蛋白体外消化率的降低。而曹云刚[22]研究氧化及没食子酸添加对MP凝胶体外消化率的影响时发现,加热前的氧化处理对MP凝胶的消化率无显著影响,上述结果不一致,可能与实验所用肉的种类、试剂的量比以及实验条件的不同有关。
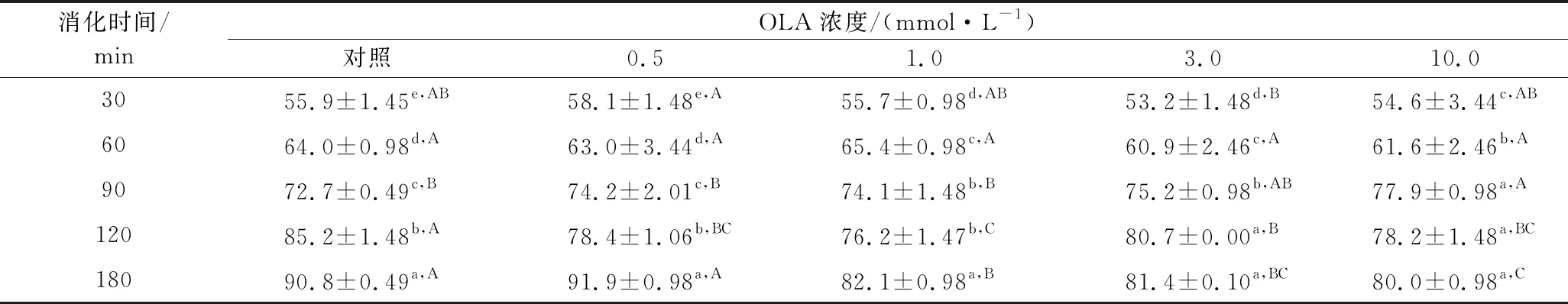
表4 不同OLA浓度下MP凝胶的体外消化率Table 4 In vitro digestibility of myofibrillar protein gels at different concentrations of OLA
3 结论
OLA诱导的蛋白氧化主要倾向于降低MP的凝胶性能,而这主要依赖于蛋白理化性质以及结构的改变。一般情况下,MP的氧化导致羰基的形成、总巯基和自由氨基含量的下降、Zeta电位绝对值的降低、蛋白质的去折叠化以及由此导致的蛋白粒径的增大和溶解度的下降。同时,轻度的氧化对MP的胶凝特性没有明显影响,而加热前MP的过度氧化会降低MP凝胶中蛋白颗粒间的疏水相互作用和氢键作用,从而降低了MP的凝胶性能以及蛋白凝胶的消化率。这些实验结果为研究OLA诱导的MP结构变化及其与凝胶性能之间的关系提供了参考。
猜你喜欢
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
现代临床医学(2021年6期)2021-11-20
食品与生物技术学报(2021年5期)2021-06-24
中国畜牧杂志(2021年5期)2021-05-17
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
昆明医科大学学报(2021年1期)2021-02-07
国际种业前沿动态(2020年16期)2020-10-14
三农资讯半月报(2020年16期)2020-09-14
湖南饲料(2019年5期)2019-10-15
