
氮掺杂Stone-Wales缺陷石墨烯吸附H2S的密度泛函理论研究
2021-10-04 15:09马生贵田博文周雨薇陈琳江霞高涛
化工学报 2021年9期
马生贵,田博文,周雨薇,陈琳,江霞,高涛
(1 四川大学建筑与环境学院,四川成都 610065;2 国家烟气脱硫工程技术研究中心,四川成都 610065;3 四川大学宜宾园区,四川宜宾 644000;4 四川大学原子与分子物理研究所,四川成都 610065)
引言
H2S 是一种具有恶臭鸡蛋气味的有毒气体,也是造成酸雨和雾霾等环境污染的主要原因[1]。其广泛地存在于各类化学工业生产过程中,对人类的健康和工业生产的安全等造成了较大的威胁。根据文献报道[2-3],过高浓度的H2S会导致人的嗅觉失灵,而低浓度的H2S也可能对人的呼吸系统和神经系统等造成损伤。此外,H2S 对金属也存在强烈的腐蚀作用,会导致金属管道的堵塞和仪器设备的腐蚀。因此,探索科学高效的H2S 脱除方法已经成为了大气污染治理的重要课题。
目前,常用的H2S废气处理方法主要包括化学吸收法、物理吸收法和吸附法三种[4]。其中,吸附法的应用最为广泛。炭基材料如活性炭是吸附法脱除H2S 最常用的吸附剂,具有孔隙结构发达、比表面积大、化学稳定性好和吸附性强等良好的物理化学性质,并因此受到科学界和工业界的青睐。Bagreev等[5]利用三种微孔活性炭作为H2S吸附剂,通过氮吸附、Boehm 滴定、电位滴定和热分析等方法对其表面性质进行了评价,发现表面化学性质、孔隙率和保水能力等因素的合理组合可以使炭具有良好的H2S吸附性能。Adib 等[6]采用尿素浸渍和热处理的方法对木质活性炭进行改性,发现活性炭经尿素改性后,H2S 向水溶性物质的转化率明显较高。然而,活性炭本身是一种无定形炭,且其化学结构比较复杂;相比而言,石墨烯是一种具有完美六边形结构的二维材料,并且还具有良好的理化性质和力学性能。因此,在基于密度泛函理论的模拟计算中,常选取石墨烯作为炭基材料的微观模型[7-9]。基于上述研究结果,为了进一步研究炭基材料对H2S 分子的吸附性能,以指导脱硫催化剂的筛选与合成,本研究以石墨烯作为微观模型,探究其与H2S 分子间的相互作用。
基于第一性原理的密度泛函理论方法是一种在材料的电子结构计算中十分常用且有效的方法[10-12]。第一性原理方法不使用任何经验公式及拟合参数作为假设,而是直接根据已知的量子力学原理从头开始计算,故而拥有其他半经验法所不可比拟的优势[13-15]。目前文献中已有大量基于密度泛函理论探究石墨烯吸附机理的报道。Leenaerts 等[16]利用第一性原理方法研究本征石墨烯对H2O、NH3、CO、NO2和NO 的吸附性能,发现了石墨烯与吸附质之间的电荷转移取决于吸附质相对于石墨烯表面的取向。Lazar 等[17]研究了丙酮等七种有机小分子在石墨烯表面的吸附焓,并利用反相气相色谱技术进行了实验验证。结果表明理论计算结果与实验测量结果符合很好,证明了密度泛函理论研究气体分子与催化剂相互作用的可靠性。
大量研究表明[18-19],本征碳材料如活性炭及完美石墨烯等对气体小分子的吸附性能并不够理想,通过构造缺陷和掺杂原子等方式对其进行结构改性,可显著增强碳材料的吸附性能。理论和实验都证明[20]石墨烯等碳材料在生长过程中通常会存在多种缺陷,如单空位缺陷、双空位缺陷、Stone-Wales(SW)缺陷等。Stone-Wales 缺陷是石墨烯分子的一对碳原子旋转而产生的[21]。碳原子对的旋转使得相邻的四个六元环转变为两个五元环和两个七元环,而这种转化也导致了电子态的局域化,增强了碳材料表面的反应活性。掺杂也是改善石墨烯等碳材料活性位点的方式之一。Dai 等[22]探讨了NO 和NO2分子在B、N、Al、S 四种不同的金属和非金属元素掺杂的石墨烯表面的吸附行为,发现B 和S 掺杂的石墨烯更易与NO和NO2发生反应,是气体传感器的良好材料。Zhou等[20]研究了甲醛分子在铬、锰、钴三种不同金属原子掺杂的SW 缺陷石墨烯表面的吸附行为,发现缺陷和掺杂都促进了甲醛在石墨烯表面的吸附。Jia 等[23]研究了锰掺杂单空位缺陷石墨烯对CO分子的吸附,发现锰原子掺杂显著地提升了石墨烯表面与CO 之间的吸附能,增加了二者间的电荷密度。这些研究都证明了缺陷和掺杂对石墨烯等碳材料和吸附质之间相互作用的影响。本文探究了H2S在氮掺杂SW 缺陷石墨烯表面的吸附行为,以说明缺陷和掺杂对石墨烯吸附作用的影响。研究结果对炭基材料脱除H2S的实验室研究及工业应用有一定的理论指导意义。
1 计算模型
1.1 计算方法
本文基于密度泛函理论方法进行吸附性质的计算,采用的计算软件是维也纳从头算模拟软件包(ViennaAb-initioSimulation Package,VASP)[24-26]。采用广义梯度近似和交换关联泛函PBE(Perdew-Burke-Ernzerhof,PBE)[27]。赝势选用投影缀加平面波方法(Projector-Augmented Wave,PAW)[28]。通过DFT-D3 描述范德华力修正[29]。结构优化的收敛标准为:作用在所有原子上的力都小于0.01 eV/Å(1 Å=0.1 nm),且收敛性判别标准为相邻两电子步之间能量差小于1×10-5eV。
为了证明所选参数的准确性和有效性,本研究对截断能(ENCUT)和k点积分网格(KPOINTS)进行了收敛性测试。其中,k点积分网格的收敛性测试的横坐标指n×n×1 中n的值。收敛性测试结果如图1所示。根据收敛性测试的结果,截断能被设置为500 eV,k点为5×5×1。在上述参数条件下,系统总能量满足收敛性,且计算耗时较少。
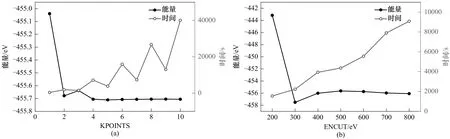
图1 能量收敛性测试:(a)k点;(b)截断能Fig.1 The energy convergence test:(a)k-points;(b)kinetic energy cutoff
在本文中,氮掺杂SW 缺陷石墨烯的形成能(Ef)计算公式如下:
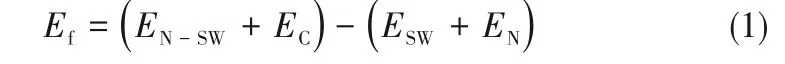
其中,EN-SW、ESW、EC、EN依次为氮掺杂SW 缺陷石墨烯表面的总能量、SW 缺陷石墨烯表面的总能量、单个碳原子的能量和单个氮原子的能量。
石墨烯表面吸附H2S 分子的吸附能(Eads)计算公式如下:
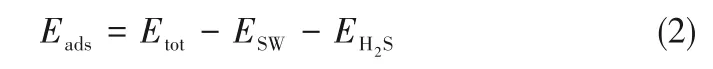
其中,Etot、ESW、EH2S依次为SW 缺陷或氮掺杂SW缺陷石墨烯吸附H2S分子后体系的能量、SW 缺陷或氮掺杂SW 缺陷石墨烯表面的能量、H2S 分子的能量。
1.2 模型选择
为了研究H2S 分子在SW 缺陷石墨烯表面的吸附情况,本研究在建立模型时选取了大小为5×5×1的石墨烯超胞,并且在z方向设置了一个20 Å 的真空层以隔绝相邻两层之间的相互作用。SW 缺陷石墨烯模型如图2所示,依据石墨烯表面的对称性,本研究选取了顶位(Top)、桥位(Bridge)、五元环穴位(Hollow-5)和七元环穴位(Hollow-7)四个典型的吸附位点研究H2S 分子在SW 缺陷石墨烯表面的吸附。同时,为了探究最稳定的氮掺杂SW 缺陷石墨烯的几何构型,本研究考虑了五种不等价的氮掺杂位点。
2 结果分析
2.1 H2S分子在SW缺陷石墨烯表面的吸附行为
H2S 分子在SW 缺陷石墨烯表面的初始构型主要有三种几何方向:H—S 键平行于SW 缺陷石墨烯的表面、硫原子接近碳原子和氢原子接近碳原子。根据图2,H2S 分子在SW 缺陷石墨烯上一共有四个吸附位点。因此,一共有12 种SW 缺陷石墨烯吸附H2S分子的初始吸附构型。本研究对12种吸附构型进行了几何优化,并计算了吸附能。表1所示为SW缺陷石墨烯吸附H2S分子的吸附能。其中,H2S分子的吸附位点同图2所示一致。
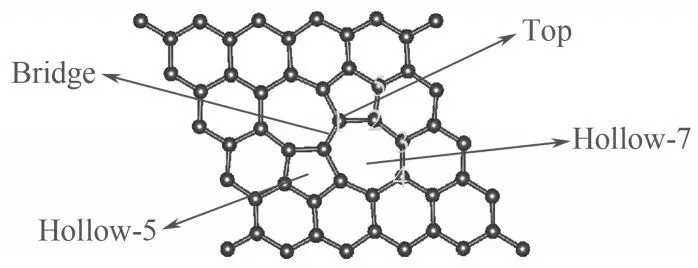
图2 SW缺陷石墨烯几何优化构型俯视图(Top、Bridge、Hollow-5和Hollow-7指H2S分子的吸附位点,数字1~5指氮原子的掺杂位点)Fig.2 The top view optimized geometric structures of SW defected graphene(Top,Bridge,Hollow-5 and Hollow-7 are the adsorption sites of H2S,number 1—5 are the doping sites of N atom)
吸附能指分子或者原子从气相中吸附到表面所释放的能量。吸附能的绝对值越大,证明气体分子被表面吸附后体系下降的能量越大,也即整个体系更加稳定。根据表1 的数据显示,H2S 分子在SW缺陷石墨烯上的吸附能绝对值显著高于其在纯净石墨烯上的吸附能(-0.064 eV)[30]。初始构型为氢原子朝下的H2S 分子在四个吸附位点的吸附能都偏低。在Top 位点,初始构型为H—S 键平行于石墨烯表面的H2S 分子具备更大的吸附能,而在Hollow-5和Bridge 位点初始构型为硫原子朝下的H2S 分子的吸附能更大。Top、Hollow-5、Bridge 三个吸附位点的最稳定构型如图3 所示。根据表1 所示的数据,只有硫原子朝下的H2S分子在Hollow-5位点上的吸附能达到了-0.51 eV,可以认为发生了弱化学吸附。其余几种情况的体系吸附能都未达到化学吸附的临界点。因此,需进一步提升SW 缺陷石墨烯对H2S分子的吸附性能。
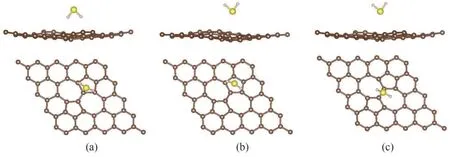
图3 H2S分子在SW缺陷石墨烯的三种吸附位点上的最稳定构型:(a)Top位点上吸附构型的侧视图和俯视图;(b)Hollow-5位点上吸附构型的侧视图和俯视图;(c)Bridge位点上吸附构型的侧视图和俯视图Fig.3 The most stable structures of H2S molecules adsorbed on the SW defected graphene at three adsorption sites:(a)side and top view of the adsorption structure at Top site;(b)side and top view of the adsorption structure at Hollow-5 site;(c)side and top view of the adsorption structure at Bridge site

表1 SW缺陷石墨烯吸附H2S分子的吸附能Table 1 Adsorption energies of H2S molecules adsorbed on the SW defected graphene
根据上文H2S 分子在SW 缺陷石墨烯表面的吸附能计算结果可知,硫原子朝下的H2S 分子在Hollow-5 位点上(下称S-Hollow-5 吸附体系)的吸附能绝对值在全部12种构型中最大,也即该构型在所有构型中最稳定。为了进一步研究该构型的电子转移情况和成键情况,计算了该构型的差分电荷密度、Bader 电荷、总态密度(density of states,DOS)和局域态密度(local density of states,LDOS)。S-Hollow-5吸附体系的电荷密度和差分电荷密度图如图4 所示,Bader 电荷数值如表2 所示。其中,表2 列出了离H2S分子最近的五个碳原子。差分电荷密度图中黄色区域表示带正电荷,蓝色区域表示带负电荷。可以看出,吸附态的H2S分子带正电荷,而石墨烯表面带负电荷。H2S 分子下方的五元碳环的五个碳原子分别有不同程度的得失电荷,而H2S 分子中的硫原子在反应中失电子,起电子供体作用。

图4 S-Hollow-5吸附体系的电荷密度(a)和差分电荷密度图(b)(等值面值:0.0001)Fig.4 Charge density(a)and deformation charge density(b)of the S-Hollow-5 adsorption system(values of absolute isosurface:0.0001)
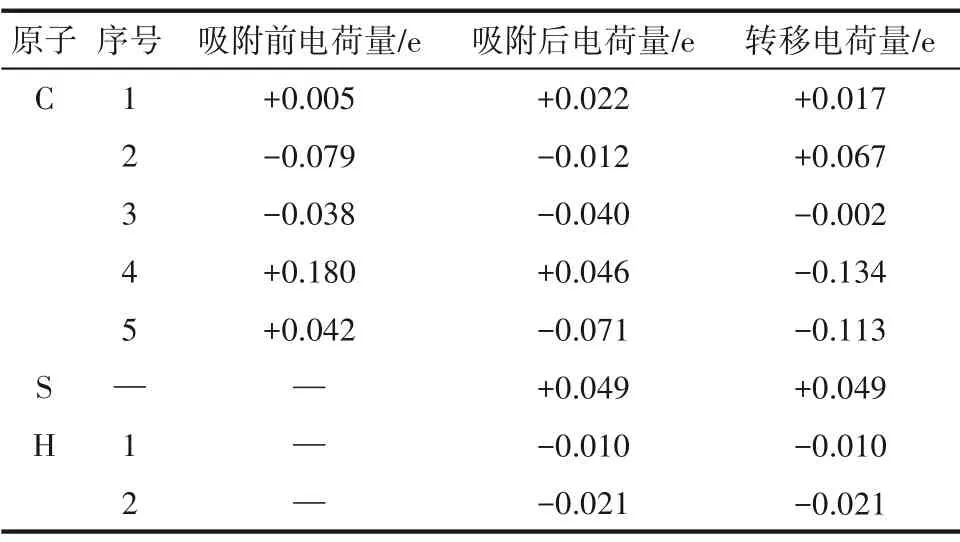
表2 S-Hollow-5吸附体系的Bader电荷Table 2 Bader atomic charges of the S-Hollow-5 adsorption system
S-Hollow-5 吸附体系的态密度和局域态密度如图5 所示。由图5 可以看出,H2S 分子的局域态密度与石墨烯表面的局域态密度的峰发生重叠,这证明SW 缺陷石墨烯表面与H2S 分子间产生了较强的相互作用。此外,H2S 分子的局域态密度主峰分别位于-14.40、-6.81、-4.96、-2.16、3.43 eV 五个能级处,且峰显得十分尖而细。这表明吸附发生后,H2S分子的外层轨道电子局域化较强,说明吸附剂表面与吸附质之间所发生的相互作用是弱相互作用,而这也与前文吸附能的计算结果相符合。综上所述,态密度和吸附能的计算结果都表明:H2S 分子与SW缺陷石墨烯表面之间发生的是物理吸附或较弱的化学吸附,SW 缺陷石墨烯并非理想的对H2S分子的吸附剂。因此,有必要通过掺杂等方式对SW 缺陷石墨烯进行改性。
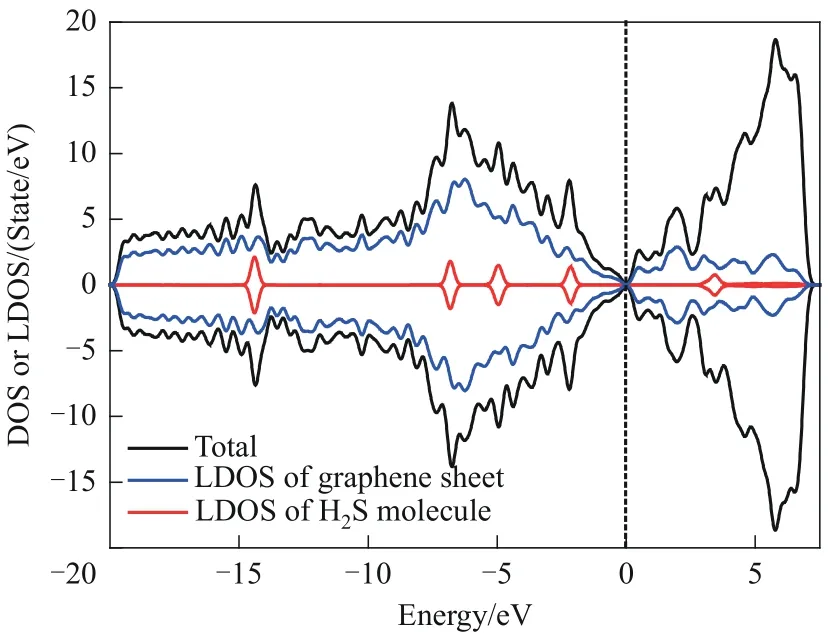
图5 S-Hollow-5吸附体系的态密度、石墨烯表面的局域态密度和H2S分子的局域态密度Fig.5 The DOS of S-Hollow-5 adsorption system,the LDOS of graphene sheet and the LDOS of H2S molecule
2.2 H2S分子在氮掺杂SW 缺陷石墨烯表面的吸附行为
根据上文所述,SW 缺陷石墨烯并非很好的对H2S 分子的吸附剂。根据文献的报道,掺杂是一种常见的改变表面性质的方法,碳原子与氮原子在元素周期表中位置相邻,原子大小相似,因此,氮原子和碳原子可以相互替换。然而,由于氮原子和碳原子的电负性不同,氮原子的掺杂可能会改变材料表面的电荷分布,从而改变吸附剂的理化性质。因此,本小节探究了H2S分子在氮掺杂SW 缺陷石墨烯表面的吸附行为,以确定具有更好吸附效果的SW缺陷石墨烯表面。
如图2 所示,本研究共选取了五种不等价的氮掺杂位点。为了探究系统在不同位点进行氮掺杂的稳定性,本研究对在五个不同位点进行氮掺杂后得到的SW 缺陷石墨烯的形成能进行了计算。氮掺杂SW缺陷石墨烯的形成能的计算结果如表3所示。

表3 氮掺杂SW缺陷石墨烯的形成能Table 3 Formation energies of N doped SW defected graphene
物质或分子构型的形成能是从参考组的分子生成该构型所吸收(或释放)的能量,形成能越低,形成体系所需要吸收的能量越少,体系也更加容易形成。根据表3 所示数据,氮原子在5 号位点掺杂时所需要的形成能最小,这意味着氮原子最容易在该位点掺杂。结合上文吸附能计算结果,H2S 分子在Hollow-5 位点上的吸附是最稳定的,而氮原子的5 号掺杂位点也恰好位于五元环上,说明五元碳环上的电荷分布可能有助于原子或分子在石墨烯表面的吸附或掺杂。
根据前文SW 缺陷石墨烯吸附H2S分子的研究,最稳定的吸附构型是硫原子朝下的H2S分子吸附在Hollow-5 位点上。因此,在氮掺杂SW 缺陷石墨烯吸附H2S 分子的研究中,初始构型为硫原子朝向石墨烯的H2S分子吸附在靠近氮原子的Hollow-5位点上的吸附体系(记为S-H 体系)仍然被作为重点进行研究。为了进一步比较吸附位点及H2S分子初始构型的影响,本文同时研究了初始构型为硫原子朝下的H2S 分子吸附在掺杂氮原子顶位的吸附体系(记为S-T体系)及初始构型为氢原子朝下的H2S分子吸附在靠近氮原子的Hollow-5 位点上的吸附体系(记为H-H 体系)。几何优化后三个体系的稳定构型如图6所示。
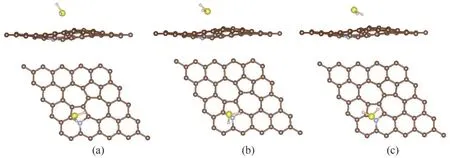
图6 H2S分子在氮掺杂SW缺陷石墨烯上的三种吸附体系的稳定构型:(a)S-H体系吸附构型的侧视图和俯视图;(b)S-T体系吸附构型的侧视图和俯视图;(c)H-H体系吸附构型的侧视图和俯视图Fig.6 The stable structures of three adsorption systems of H2S molecules adsorbed on the N doped SW defected graphene:(a)side and top view of the adsorption structure of S-H system;(b)side and top view of the adsorption structure of S-T system;(c)side and top view of the adsorption structure of H-H system
为了探究氮掺杂后SW 缺陷石墨烯吸附H2S 分子的稳定性,本文比较了三种吸附体系的吸附能(表4)。结果发现,三种吸附体系的吸附能绝对值都大于0.50 eV,说明三种体系中都产生了化学吸附。这与前文H2S 分子在SW 缺陷石墨烯上的吸附以物理吸附为主的情况相比发生了较大的变化,更强的相互作用在氮掺杂后的石墨烯表面与H2S分子之间发生。通过比较三种吸附体系的吸附能,发现三个体系的吸附能之间的差值很小,最大仅0.09 eV,这证明在含掺杂氮原子的五元环附近,吸附位点和分子取向所造成的影响减小,也证明了氮原子的掺杂对吸附作用的影响。

表4 氮掺杂SW缺陷石墨烯吸附H2S分子的吸附能Table 4 Adsorption energies of H2S molecules adsorbed on the N doped SW defected graphene
为了进一步探究氮原子掺杂对吸附作用的影响,本文进行了差分电荷、Bader 电荷、态密度和局域态密度的分析研究。和上文一样,本小节的研究选取了吸附能最大的体系,即H-H 吸附体系为例进行研究。H-H 吸附体系的电荷密度及差分电荷密度图如图7 所示,Bader 电荷值如表5 所示。电荷分析的结果显示:氮原子掺杂后,H2S 分子在吸附反应过程中的电荷转移量为0.028 e,根据表2结果,对于SW 缺陷石墨烯,H2S 分子在吸附反应过程中的电荷转移量仅为0.018 e。对比二者数据可以发现,氮原子掺杂后,H2S 分子的电荷转移量有了显著提高,这证明氮原子的存在促进了石墨烯表面与H2S分子间的电荷转移进程,强化了二者的相互作用。是由于氮原子的掺杂改变了石墨烯表面的电荷分布。在未吸附H2S 分子时,掺杂氮原子的最外层电子数增加了1.283 e,而周围的碳原子则因失电子而带有正电荷。进一步分析发现,在吸附反应过程中,氮原子所带电荷量几乎保持不变,这说明氮原子主要作为电子传递的桥梁参与H2S与石墨烯表面之间的电荷转移。
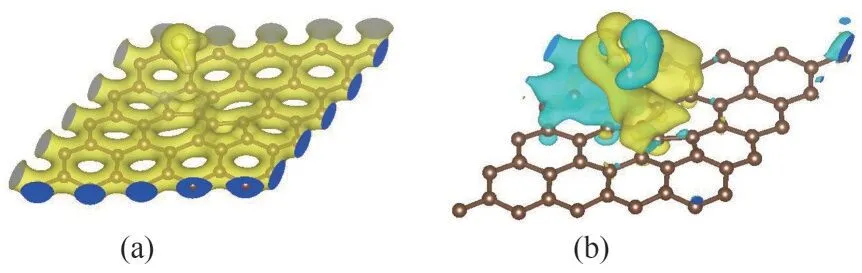
图7 H-H吸附体系的电荷密度(a)和差分电荷密度图(b)(等值面值:0.0001)Fig.7 Charge density(a)and deformation charge density(b)of the H-H adsorption system(values of absolute isosurface:0.0001)
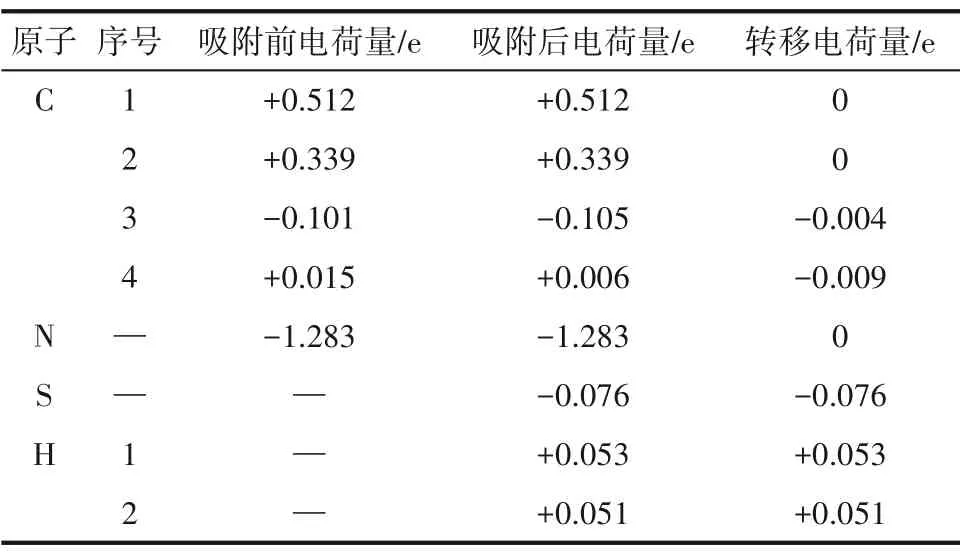
表5 H-H吸附体系的Bader电荷Table 5 Bader atomic charges of the H-H adsorption system
H-H吸附体系的态密度结果如图8所示。对比图8 与图5 的结果发现:H2S 分子的局域态密度的峰在氮原子掺杂后变得矮而宽,这证明在氮原子掺杂后,H2S 分子的外层电子的能级分布变得更广,电子离域化加强。一般而言,当吸附质与吸附剂表面发生化学反应后,吸附质外层电子的离域化会增强,进一步证明了H2S 分子与氮掺杂SW 缺陷石墨烯表面之间发生的是化学吸附。然而,与未掺氮SW 缺陷石墨烯和H2S 分子的吸附体系相比,掺氮后的吸附体系的态密度图形并没有发生大的变化,说明二者之间发生微弱的化学吸附。在未来的研究中,或许可以尝试通过掺杂多个氮原子或者掺杂其他原子的方式来更进一步地改善SW 缺陷石墨烯表面的性质。
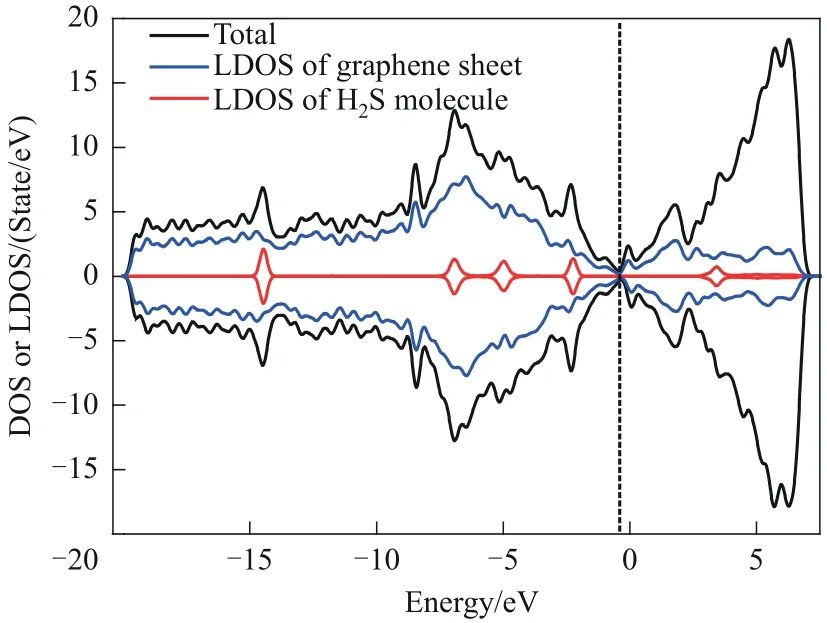
图8 H-H吸附体系的态密度、石墨烯表面的局域态密度和H2S分子的局域态密度Fig.8 The DOS of H-H adsorption system,the LDOS of graphene sheet and the LDOS of H2S molecule
3 结论
(1)H2S 分子在SW 缺陷石墨烯上的吸附以物理吸附为主,H2S分子在氮掺杂SW 缺陷石墨烯上吸附的最稳定构型为较弱的化学吸附。
(2)氮掺杂可以有效提升SW 缺陷石墨烯对H2S分子的吸附能,加强了石墨烯表面与H2S 分子之间的相互作用。
(3) 在SW 缺陷及氮掺杂SW 缺陷石墨烯吸附H2S 的体系中,H2S 分子主要作为电子供体,而石墨烯表面主要作为电子受体。此外,氮原子的引入改变了石墨烯表面的电荷分布,使其周围的碳原子因失电子而带有正电荷。氮原子主要作为电子传递的桥梁参与H2S与石墨烯表面之间的电荷转移。
(4)H2S 分子通常在SW 缺陷石墨烯的五元碳环中心这一位点上的吸附具有最大的吸附能绝对值,这证明五元碳环上的电荷分布有助于原子或分子在石墨烯表面的吸附或掺杂。
猜你喜欢
舰船科学技术(2022年21期)2022-12-12
物理学报(2022年11期)2022-06-18
军民两用技术与产品(2022年1期)2022-06-01
山西大学学报(自然科学版)(2021年5期)2021-12-25
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
物理学报(2019年23期)2019-12-16
北京航空航天大学学报(2017年10期)2017-04-20
新高考·高一物理(2015年6期)2015-09-28
