
大环芳烃菲咯啉的设计、合成及其铜光敏剂光解水制氢性能
2021-09-22 02:10俞晓聪刘雪粉范金轩王天琦吴庆安罗书平
无机化学学报 2021年9期
许 萌 俞晓聪 刘雪粉 陈 浩 范金轩 王天琦 吴庆安 罗书平*,
(1浙江工业大学绿色化学合成技术国家重点实验室培育基地,杭州 310014)
(2杭州师范大学钱江学院,杭州 310018)
随着经济的快速发展,能源和环境成了人们日益关注的话题。特别是人类对能源的需求量日益增长,而石油等不可再生资源即将枯竭,寻找可再生的清洁能源迫在眉睫[1‑3]。氢能源被认为是21世纪最具发展潜力的绿色能源,甚至被称为“人类的终极能源”,人们对氢能的开发应用寄予了极大的热忱和希望[4‑5]。太阳能是当前广泛使用的清洁能源。利用太阳能光解水将光能转化为氢气能源是一种有前景的方法,有望应对能源危机和解决日益恶化的环境问题[6‑7]。
光催化分解水反应可以分为2个半反应:水氧化生成氧气和水还原生成氢气[8‑10]。为了研究水还原中光解水制氢的反应机理及催化体系,通常加入过量的电子牺牲剂,可屏蔽水氧化半反应的影响。目前水还原制氢系统主要由光敏剂、水还原催化剂和电子牺牲剂组成[11]。由于光敏剂在高效光子捕获、激发态电子的产生和转移过程中的作用是极其重要的,因此它是制约光催化分解水产氢效率的关键组分。自1970年首次报道了[Ru(bpy)3]2+可以作为光敏剂[12‑13],随后科学家们又研发了一系列其他贵金属光敏剂,比如铱[14‑16]、铂[17‑19]、铑[20‑21]等。但由于其昂贵的价格、稀缺的资源和污染环境等缺点限制了它们在光解水制氢体系中的应用。从2012年起,我们课题组着重对非贵金属的氮磷杂配铜光敏剂进行了研究,发现对氮配体1,10‑菲咯啉进行结构改造会对铜基光敏剂的光敏活性产生非常重要的影响[22‑25]。
为了研究氮配体菲咯啉4,7位取代基对光敏剂活性的影响,我们设计、合成了一系列4,7位为大环芳烃取代的2,9‑二甲基‑1,10‑菲咯啉(图1),研究其在杂配铜光敏剂光解水制氢中的作用和构效关系以及光物理化学性能。
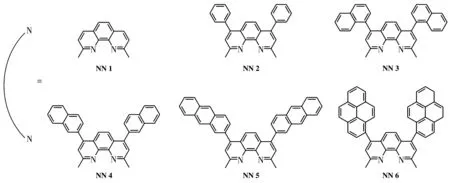
图1 4,7‑二芳基‑2,9‑二甲基‑1,10‑菲咯啉衍生物Fig.1 4,7‑diaryl‑2,9‑dimethyl‑1,10‑phenanthroline ligand derivatives
1 实验部分
1.1 仪器与试剂
底物4,7‑二溴‑2,9‑二甲基‑1,10‑菲咯啉是参考文献[26‑27]制得,其余底物及试剂均购自商业公司,所有溶剂都经过重蒸除水。NMR分析使用瑞士Bruker公司的AdvanceⅢ核磁共振仪;紫外分析使用瓦里安公司的CARY100紫外可见近红外分光光度计;荧光分析使用SHIMADZU公司的RF‑6000荧光分析仪;循环伏安(CV)分析使用上海辰华的CHI电化学工作站;MS分析使用美国Thermo公司LCQ advantage质谱仪;HR‑MS分析使用美国Agilent公司的Agilent 6210 TOF LC/MS。
1.2 合 成
1.2.1 4,7‑二芳基‑1,10‑菲咯啉衍生物及其合成
4,7‑二芳基‑1,10‑菲咯啉衍生物及其合成方法如图2所示。

图2 4,7‑二芳基‑1,10‑菲咯啉配体NN 3~NN 6的合成方法Fig.2 Synthetic method of 4,7‑diaryl‑1,10‑phenanthroline ligands NN 3~NN 6
NN 3配体的合成:在25 mL三口烧瓶中依次加入4,7‑二溴‑2,9‑二甲基‑1,10‑菲咯啉(0.36 g,1 mmol)、1‑萘硼酸(0.36 g,2.1 mmol)、碳酸钾(0.69 g,5 mmol)和Pb(PPh3)4(0.058 g,0.05 mmol),用氮气抽换气3次,再加入9 mL混合溶剂(V甲苯∶V乙醇∶V水=1∶1∶1),用氮气鼓泡0.5 h后,搅拌升温至85℃,反应12 h后,溶液变为棕黑色。TLC跟踪,待原料反应完全后,冷却至室温。将反应液加入50 mL水,用60 mL二氯甲烷萃取3次,合并有机相,再用饱和食盐水洗2次,干燥,柱层析提纯(V二氯甲烷/V乙酸乙酯=3/2),之后再用二氯甲烷和正己烷重结晶,得到淡黄色固体0.21 g,收率45%。1H NMR(400 MHz,CDCl3):δ 7.91(dd,J=11.4,8.3 Hz,4H),7.69~7.64(m,2H),7.54(d,J=5.4 Hz,4H),7.45~7.40(m,5H),7.36~7.27(m,1H),7.09(d,J=12.6 Hz,2H),3.05(s,6H)。13C NMR(100 MHz,CDCl3):δ 158.96,158.85,147.46,145.65,145.58,135.82,135.76,133.43,133.38,133.08,132.15,132.05,131.94,131.84,128.73,128.44,127.40,126.51,126.12,125.88,125.22,125.05,125.00,123.47,26.00。ESI‑MS(m/z):[M+H]+461.2。
NN 4配体的合成方法参照配体NN 3。配体NN 4:淡黄色固体,收率 53%。1H NMR(600 MHz,CDCl3):δ 8.06~7.98(m,2H),7.98~7.90(m,2H),7.82(s,2H),7.73~7.64(m,4H),7.61~7.54(m,8H),7.52~7.46(m,2H)。13C NMR(151 MHz,CDCl3):δ 158.81,148.61,145.99,135.73,133.23,132.97,132.88,132.19,132.14,132.07,131.95,131.93,128.86,128.55,128.47,128.19,128.14,127.81,127.45,126.69,126.67,124.89,124.28,123.15,25.99。ESI‑MS(m/z):[M+H]+461.2。
NN 5配体的合成方法参照配体NN 3。配体NN 5:黄色固体,收率 40%;1H NMR(400 MHz,CDCl3):δ 8.48(d,J=5.9 Hz,3H),8.33(h,J=4.9,4.2 Hz,1H),8.18~8.09(m,2H),8.06~7.98(m,2H),7.92~7.77(m,2H),7.66(dt,J=17.6,5.9 Hz,5H),7.54~7.42(m,7H),3.05(s,6H)。13C NMR(100 MHz,CDCl3):δ 158.87,149.00,146.56,146.08,145.87,145.62,135.19,134.45,133.72,132.17,131.96,131.26,130.93,129.23,128.58,128.40,128.22,127.41,127.04,126.86,126.31,125.86,124.38,124.05,123.31,25.76。ESI‑MS(m/z):[M+H]+561.2。
NN 6配体的合成方法参照配体NN 3。配体NN 6:黄色固体,收率 42%;1H NMR(400 MHz,CDCl3):δ 8.23~8.16(m,4H),8.12~8.07(m,6H),8.01~7.84(m,6H),7.69~7.52(m,4H),7.12(dd,J=13.7,2.0 Hz,2H),3.13(d,J=2.1 Hz,6H)。13C NMR(100 MHz,CDCl3):δ 158.97,158.88,148.00,145.66,132.84,131.38,131.31,130.78,129.20,128.16,127.96,127.53,127.26,126.40,126.23,125.67,125.62,125.56,125.36,124.83,124.65,124.60,124.55,124.51,124.43,123.74,123.70,26.03。ESI‑MS(m/z):[M+H]+609.2。1.2.2 铜配合物的原位合成
参考课题组之前的工作,采用原位配位的方法合成相应的铜配合物[28]。以配合物CuPS 1为例:氩气保护下,将配体 2,9‑二甲基‑1,10‑菲咯啉(1 mmol)、膦配体Xantphos(1 mmol)和六氟磷酸四乙腈合铜(1 mmol)溶于10 mL四氢呋喃中,室温下搅拌0.5 h,除掉溶剂即可得到相应的氮磷杂配铜配合物CuPS 1(图3),可直接应用于光催化分解水制氢反应。
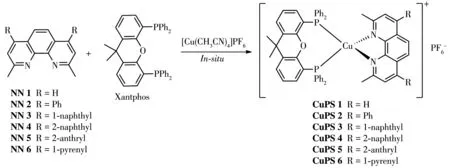
图3 铜配合物的原位合成方法Fig.3 In⁃situ synthetic method of the copper complexes
氮磷杂配铜基光敏剂CuPS 2~CuPS 6也用同样的方法获得,并通过ESI‑MS检测确认。
CuPS 1:1H NMR(600 MHz,CDCl3):δ 8.34(d,J=8.2 Hz,2H),7.83(s,2H),7.67(dd,J=7.8,1.4 Hz,2H),7.55(d,J=8.2 Hz,2H),7.27~7.19(m,6H),7.04(dq,J=5.4,3.1 Hz,16H),6.90(dtd,J=7.5,3.7,1.3 Hz,2H),2.27(s,6H),1.77(s,6H)。13C NMR(151 MHz,CDCl3):δ 158.46,155.07,142.82,137.93,133.80,132.88,131.45,131.34,129.97,128.61,127.44,126.04,125.27,121.57,36.12,28.45,27.11。ESI‑MS(m/z):[C53H44CuN2OP2+H]+850.2,[C53H44CuN2OP2+Na]+872.2。
CuPS 2:1H NMR(600 MHz,CDCl3):δ 7.80(s,2H),7.69(dd,J=7.8,1.4 Hz,2H),7.63~7.53(m,6H),7.52~7.45(m,6H),7.33~7.27(m,6H),7.10(qd,J=7.6,4.4 Hz,16H),6.99(dtd,J=7.5,3.7,1.4 Hz,2H),2.36(s,6H),1.78(s,6H)。13C NMR(151 MHz,CDCl3):δ 157.82,155.01,150.19,143.62,136.32,133.78,132.99,131.42,130.33,130.01,129.41,129.03,128.62,127.60,125.63,125.39,123.57,36.12,28.59,27.38。ESI‑MS(m/z):[C65H52CuN2OP2+H]+1 002.3,[C65H52CuN2OP2+Na]+1 024.3。
CuPS 3:1H NMR(600 MHz,CDCl3):δ 7.98(ddt,J=27.0,8.3,1.4 Hz,4H),7.72(ddt,J=7.7,3.0,1.6 Hz,2H),7.62(ddd,J=8.3,7.0,3.1 Hz,2H),7.56~7.51(m,4H),7.45~7.33(m,8H),7.31~7.12(m,22H),7.02(dqd,J=7.6,3.4,1.7 Hz,2H),2.42(s,3H),1.79(s,3H)。13C NMR(151 MHz,CDCl3):δ 158.18,155.07,149.39,143.37,133.71,133.36,132.87,132.04,131.18,130.35,130.13,129.71,128.75,127.65,126.85,126.50,125.38,125.02,124.13,36.17,28.54,27.39。ESI‑MS(m/z):[C73H56CuN2OP2+H]+1 102.3,[C73H56CuN2OP2+Na]+1 124.3。
CuPS 4:1H NMR(600 MHz,CDCl3):δ 8.05(d,J=8.4 Hz,2H),8.01(d,J=1.7 Hz,2H),7.97(ddd,J=8.1,6.3,3.7 Hz,4H),7.86(s,2H),7.70(dd,J=7.8,1.5 Hz,2H),7.63~7.58(m,8H),7.34~7.30(m,4H),7.26(t,J=7.7 Hz,2H),7.18~7.10(m,16H),7.01(dtd,J=7.5,3.6,1.4 Hz,2H),2.40(s,6H),1.79(s,6H)。13C NMR(151 MHz,CDCl3)δ 157.93,155.03,150.19,143.72,133.77,133.29,133.03,131.46,130.37,130.04,129.10,128.66,128.39,127.89,127.61,127.14,126.68,125.71,125.47,123.73,36.14,28.59,27.42。ESI‑MS(m/z):[C73H56CuN2OP2+H]+1102.3,[C73H56CuN2OP2+Na]+1 124.3。
CuPS 5:1H NMR(600 MHz,CDCl3):δ 8.24~8.17(m,4H),8.10~8.05(m,4H),7.94(s,2H),7.71(dd,J=7.9,1.4 Hz,2H),7.64(s,2H),7.58(dd,J=8.6,1.7 Hz,2H),7.55~7.51(m,4H),7.36~7.32(m,4H),7.20~7.08(m,20H),7.02(dqd,J=7.0,3.5,1.3 Hz,4H),2.42(s,6H),1.85~1.73(m,6H)。13C NMR(151 MHz,CDCl3):δ 157.93,155.04,150.20,143.80,133.82,133.05,132.51,131.47,130.99,130.39,130.07,129.64,129.22,128.68,128.32,127.61,127.29,126.49,126.10,125.62,123.80,36.14,28.61,27.44。ESI‑MS(m/z):[C81H60CuN2OP2+H]+1 202.4,[C81H60CuN2OP2+Na]+1 224.4。
CuPS 6:1H NMR(600 MHz,CDCl3):δ 8.29(dd,J=7.9,1.9 Hz,2H),8.27~8.23(m,2H),8.20(d,J=7.5 Hz,2H),8.14(qd,J=9.0,2.9 Hz,4H),8.07~7.99(m,4H),7.92(dd,J=16.9,7.8 Hz,2H),7.73(dtd,J=8.0,4.1,1.4 Hz,2H),7.68(d,J=2.0 Hz,2H),7.50~7.36(m,6H),7.33~7.17(m,20H),7.12~7.04(m,2H),2.49(s,6H),1.80(s,6H)。13C NMR(151 MHz,CDCl3):δ 158.13,155.14,149.89,143.56,133.90,133.41,132.94,131.96,131.29,130.40,128.82,127.68,127.22,126.60,126.15,125.77,124.81,124.41,123.89,36.18,34.46,28.57,27.50,23.30。ESI‑MS(m/z):[C85H62CuN2OP2+H]+1 252.9,[C85H62CuN2OP2+Na]+1 274.9。
1.3 光解水制氢
光解水制氢的一般方法:在氩气保护下,称取3.5 μmol光敏剂CuPS 1~CuPS 6加入特殊反应瓶中,再加入10 mL混合溶剂(V四氢呋喃∶V三乙胺∶V水=4∶3∶1),用循环恒温水浴槽将整个反应系统的温度稳定在25℃,搅拌1~2 min后,反应液的颜色逐渐变成淡黄色,加催化剂十二羰基三铁(5 μmol),打开300 W氙灯光源照射反应瓶,并用U形管收集产生的氢气。待反应体系中不产生气体时,停止搅拌,关闭光源,记录产生的气体体积,并用气相色谱检测收集的气体。
1.4 光谱测试
取 3.5 μmol铜基光敏剂 CuPS 1~CuPS 6,用四氢呋喃溶解后定容到50 mL容量瓶中,在室温下进行光化学测试。
1.5 CV测试
配制浓度为0.1 mol·L-1的四丁基六氟磷酸铵的乙腈溶液,加入3.5 μmol的铜基光敏剂CuPS 1~CuPS 6,定容到100 mL容量瓶中,用三电极(工作电极、对电极和参比电极)方法进行测试。所用电极:玻碳电极(d=2 mm)为工作电极;铂电极为对电极;Ag/AgNO3为参比电极。其他条件:室温,电解质是0.1 mol·L-1四丁基六氟磷酸铵的四氢呋喃溶液,扫描速度100 mV·s-1,阶跃电位2.5 mV,调制振幅25 mV,调制时间0.05 s,间隔时间0.5 s,最高电位2.0 V,最低电位-2.5 V。
1.6 理论计算
为了深入了解配体对铜基光敏剂的结构和性质的影响,同时简化计算方法,只对氮配体NN 2~NN 6进行了密度泛函理论(DFT)计算,在B3LYP/6‑31G(d)水平上进行[29]。
2 结果与讨论
2.1 紫外和荧光表征
由图4可知,铜基配合物CuPS 1~CuPS 6在300~350 nm之间有很强的紫外吸收,这归因于菲咯啉配体分子内的π‑π*电荷转移,而在380~410 nm之间较强的紫外吸收是铜基配合物的特征吸收峰——金属到配体的电子转移(MLCT)[30]。不同的是,CuPS 6在350 nm左右有一个很强的吸收峰,这是由于芘基π‑π*自身相互作用的结果[31]。由表1和图4可知,从CuPS 1到CuPS 5,随着4,7位大环芳烃基团的增大,配合物的吸收峰也逐渐发生红移(从381 nm逐步红移至406 nm),吸收强度也相应增大,相应的摩尔吸光系数也逐渐增大,从3 111 L·mol-1·cm-1增大至10 304 L·mol-1·cm-1。表明氮配体的4,7位大环芳烃取代基可显著影响铜基配合物的吸光性能,随着取代基的芳香环增大,铜配合物吸收峰红移,其摩尔吸光系数急剧增加。铜基配合物CuPS 1~CuPS 6的荧光发射谱如图5所示。可以看出,从CuPS 1到CuPS 3,由于大环芳烃基团的增大,配合物发射峰发生红移,荧光强度也逐渐增加。但是,相比于CuPS 1~CuPS 4,CuPS 5发射峰发生蓝移,荧光强度大大减弱;CuPS 6发射峰也发生蓝移,而荧光强度变化不大。根据文献[32]:芘基和蒽基是优良的电子给体,是大π电子,容易发生π‑π堆积,使分子间作用力增强,形成π聚焦或激基复合物,发生蓝移和荧光猝灭;分子吸收光子后从弱极性的基态跃迁到中等极性的荧光发射态,随后进一步发生从给体到受体的分子内电荷转移并伴有分子平面的扭曲,形成极性更大的弱荧光发射态(TICT态);芘本身是一个相当好的蓝光发色团,作为一个典型的稠环芳烃,具有独特的荧光性质,所以CuPS 6仍然具有较高的荧光强度。此外,还对它们的荧光量子产率和激发态寿命做了计算(表1),结果表明CuPS 3具有较高的荧光量子产率和较长的激发态寿命。
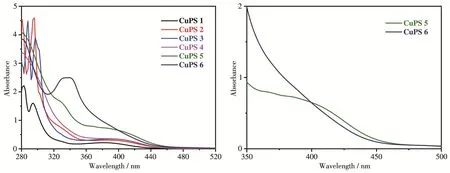
图4 CuPS 1~CuPS 6的UV‑Vis吸收谱图(左);CuPS 5和CuPS 6的局部放大UV‑Vis吸收谱图(右)Fig.4 UV‑Vis absorption spectra of CuPS 1~CuPS 6(left);Enlarged partial UV‑Vis absorption spectra of CuPS 5 and CuPS 6(right)
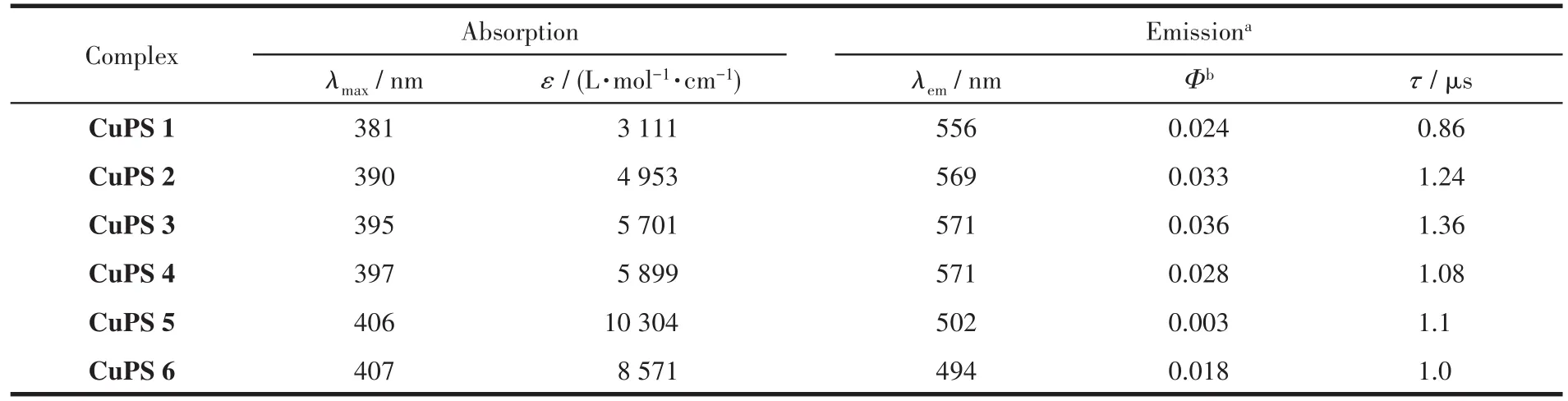
表1 CuPS 1~CuPS 6的光物理性质Table 1 Photophysical properties of CuPS 1~CuPS 6
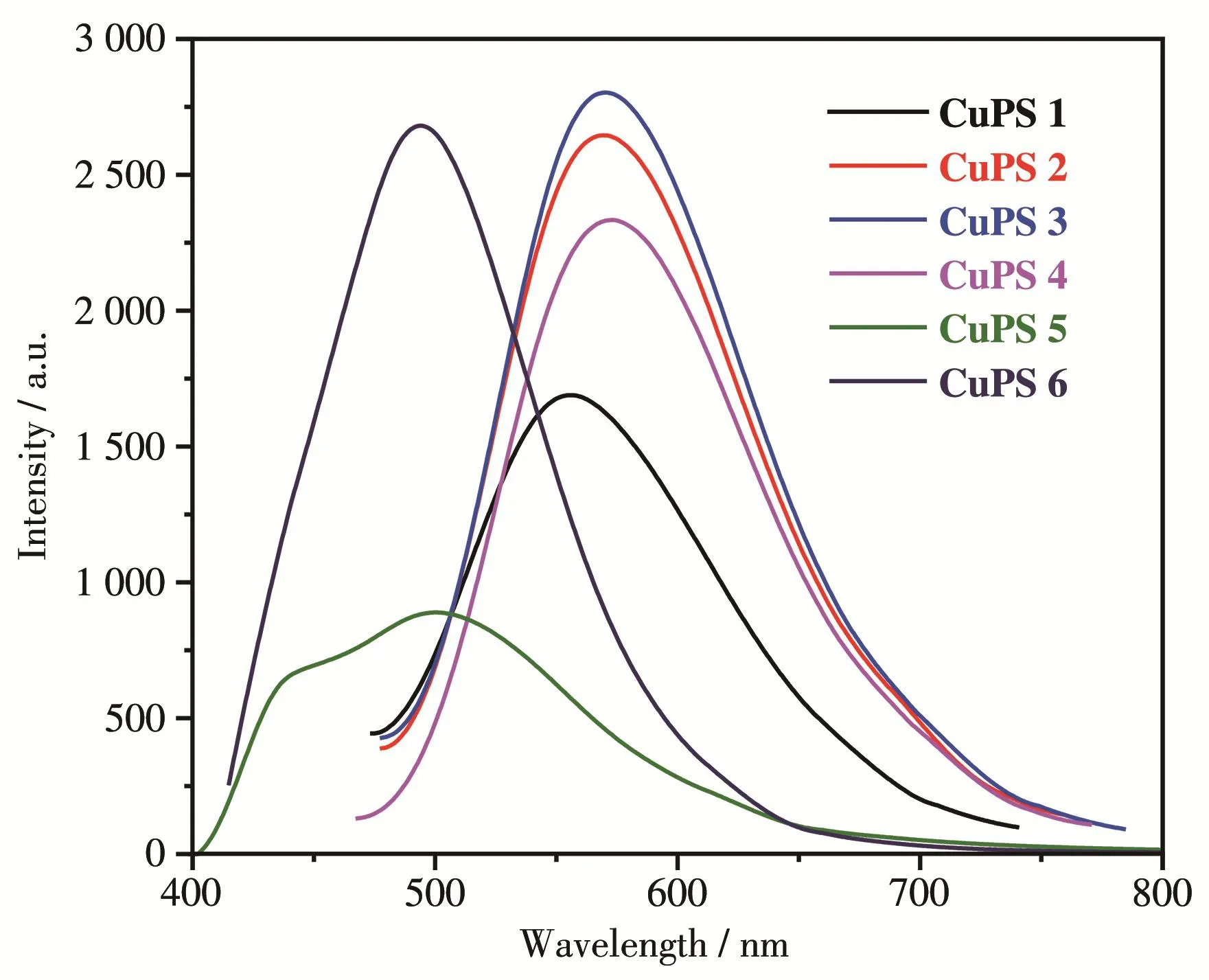
图5 CuPS 1~CuPS 6的荧光发射谱图Fig.5 Fluorescent emission spectra of CuPS 1~CuPS 6
2.2 CV表征
由图6和表2可知,所测的铜基配合物CuPS 1~CuPS 6都有一个大体相似的电位值,整个过程是一个单电子的氧化还原过程。通过对比发现,菲咯啉4、7位上的不同大环芳烃基团对于其氧化电位的影响较小,这和文献中研究结果一致[33]。相比2‑萘基取代的CuPS 4,1‑萘基取代的CuPS 3具有更强的还原能力,这与1、2‑萘基取代的氮配体NN 3和NN 4的氧化还原电位结果一致[34]。
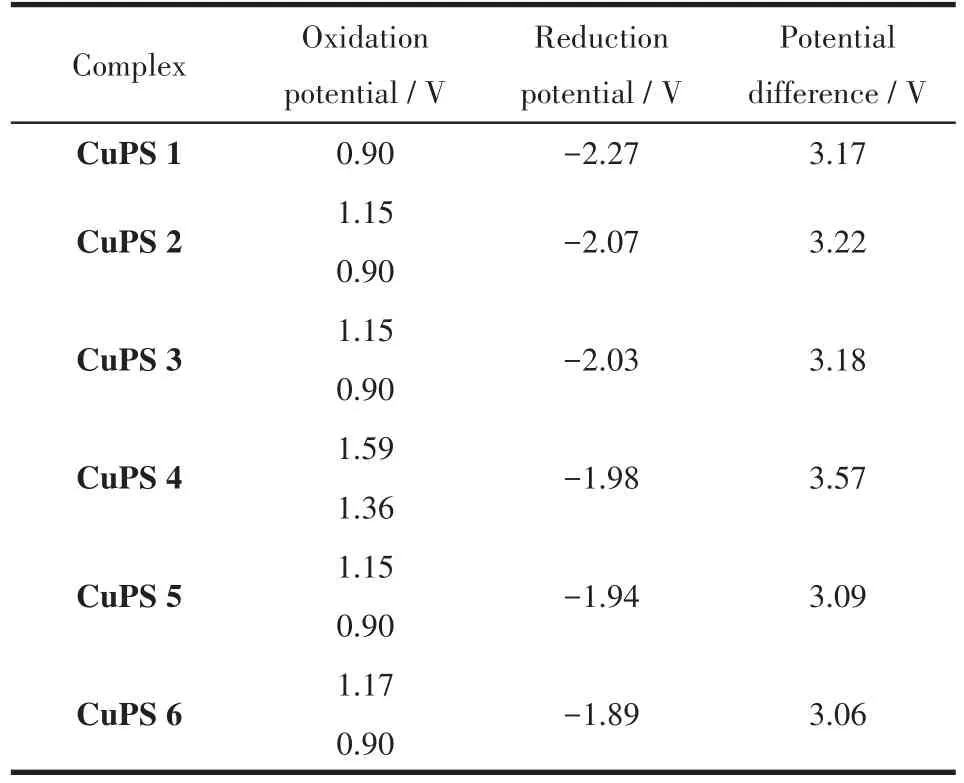
表2 CuPS 1~CuPS 6的电化学数据Table 2 Electrochemical data of CuPS 1~CuPS 6

图6 CuPS 1~CuPS 6的CV曲线Fig.6 CV curves of CuPS 1~CuPS 6
2.3 理论计算结果
由表3和图7、8可知,氮配体NN 2~NN 4的HOMO和LUMO电子云密度分布比较均匀,而NN 5~NN 6的HOMO和LUMO电子云密度主要分布在菲咯啉4,7位的取代基上。氮配体NN 3和NN 6具有较大的二面角,有利于结构的稳定性。

表3 NN 2~NN 6的理论计算结果Table 3 Theoretical calculation results of NN 2~NN 6

图7 NN 2~NN 6的NBO电荷分布图和分子空间结构图Fig.7 Calculated NBO charge distributions and molecular structures of NN 2~NN 6
2.4 制氢性能研究
由表4可以看出,不同大环芳烃基团的铜光敏剂对光解水制氢体系活性有很大的影响,这可能是因为大环芳烃基团有利于提高铜基光敏剂在制氢体系中的结构稳定性,从而提升了光敏剂的制氢活性。光敏剂CuPS 1~CuPS 6中1‑萘基取代的CuPS 3表现出最好的制氢结果,TON达957。由图7的理论计算数据可知,CuPS3与CuPS4的氮配体相比,1位取代的大环芳烃与菲咯啉母核具有更大的二面角,结构更加稳定,其荧光不易被周围环境猝灭,有利于提高制氢效果。蒽基和芘基是较强的给电子基团,但是由于其自身π‑π堆积的影响,使其平面与菲咯啉平面发生扭曲,不利于电子的传输,从而导致CuPS 5和CuPS 6的制氢性能较差。
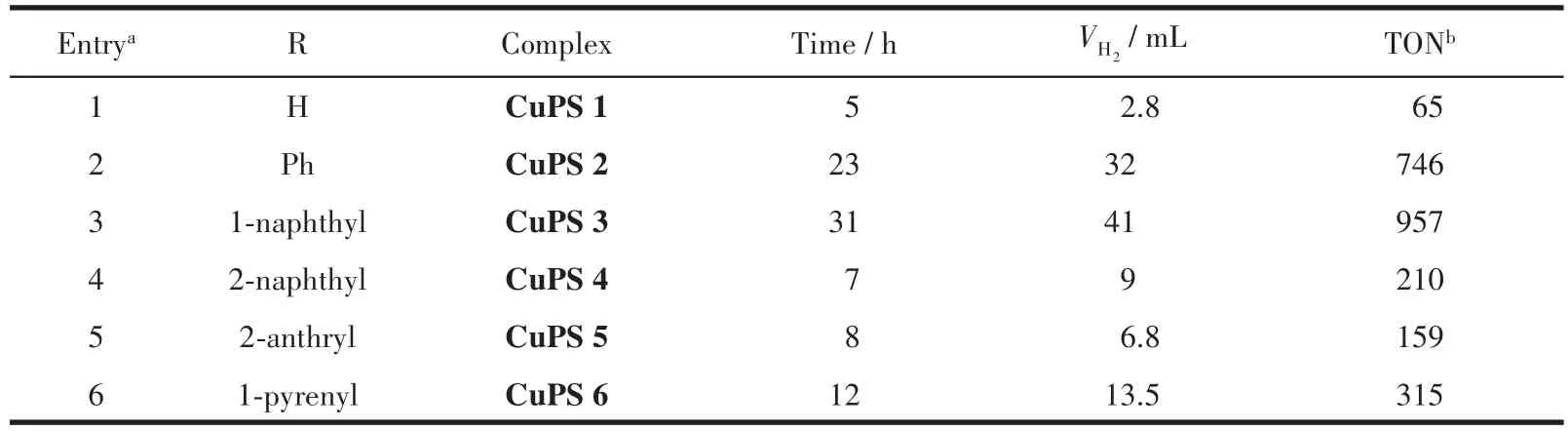
表4 CuPS 1~CuPS 6的产氢数据Table 4 Hydrogen production data of CuPS 1~CuPS 6
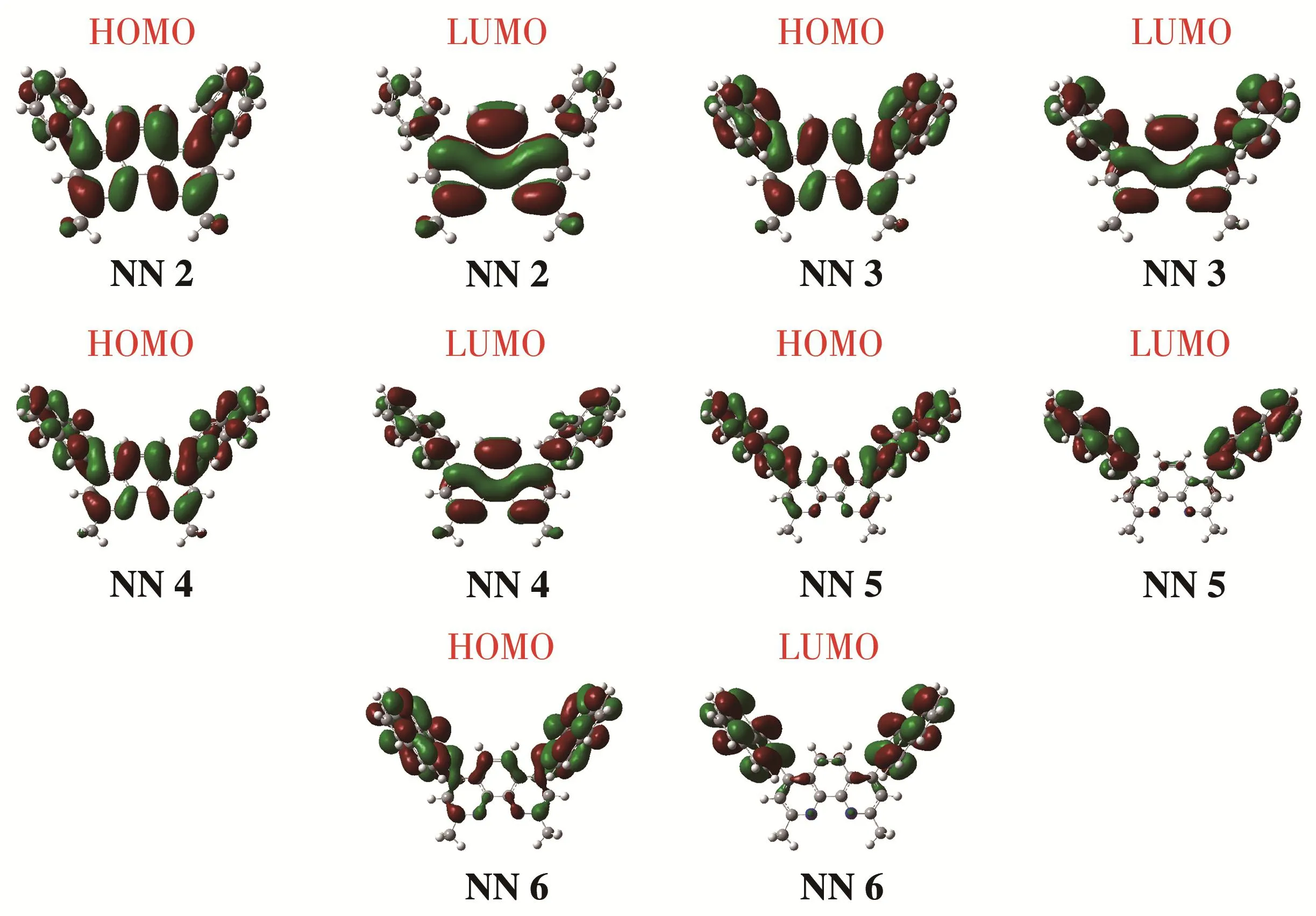
图8 NN 2~NN 6的前沿分子轨道Fig.8 Frontier molecular orbitals of NN 2~NN 6
为了确定铜基光敏剂的激发态在光解水制氢体系中的猝灭途径,我们进行了荧光猝灭实验(图9)。取3.5 μmol CuPS 3溶解在50 mL的THF中,分别加入不同浓度的TEA和Fe3(CO)12,在室温条件下测试体系的荧光强度。当TEA作为猝灭剂时,TEA的浓度几乎不影响CuPS 3的荧光强度。当Fe3(CO)12作为猝灭剂时,随着Fe3(CO)12浓度的增大,荧光强度明显下降,原因可能是加入的Fe3(CO)12与铜基光敏剂的激发态之间发生了分子间的能量转移,说明该光解水制氢体系可能为氧化猝灭途径。
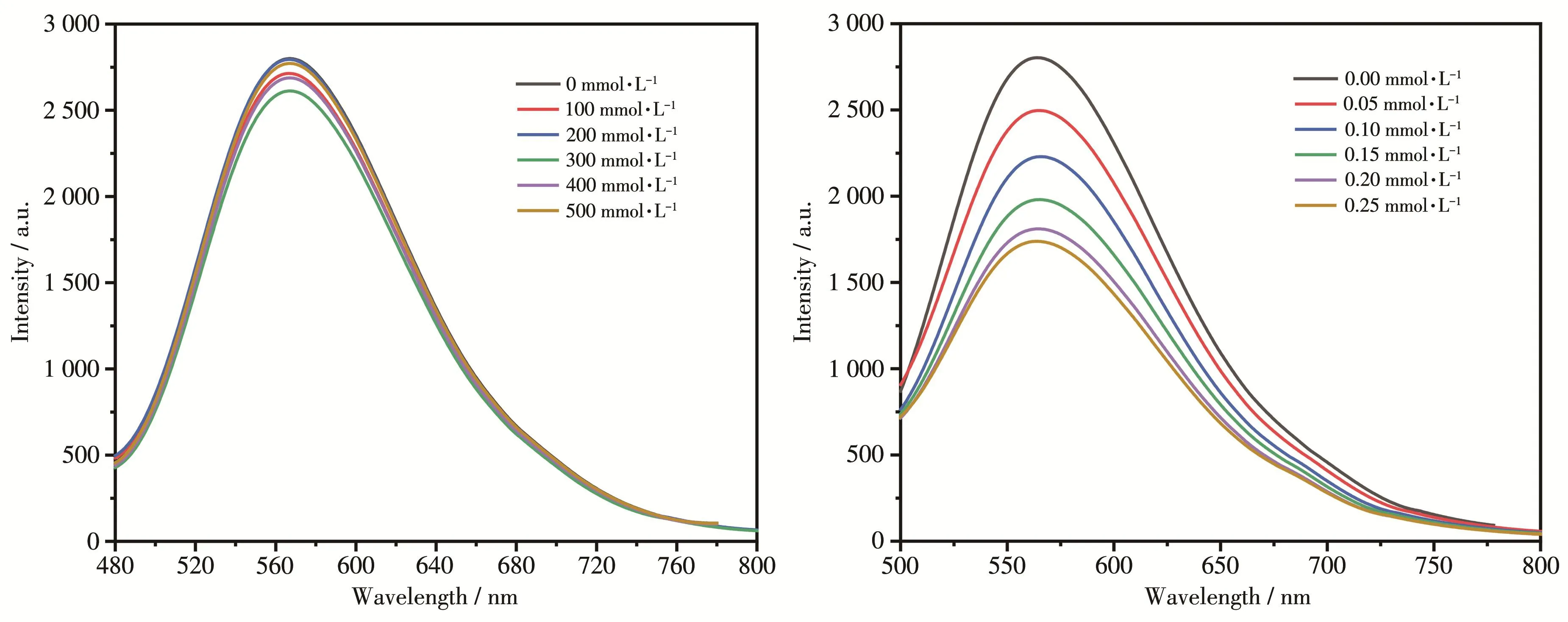
图9 不同浓度TEA(左)和Fe3(CO)12(右)对光敏剂CuPS 3的荧光猝灭Fig.9 Fluorescence spectra of CuPS 3 quenched by TEA(left)and Fe3(CO)12(right)with different concentrations
3 结论
设计、合成了一类大环芳烃菲咯啉配体(NN 1~NN 6),再与六氟磷酸四乙腈合铜和Xantphos反应,原位合成了不同大环芳烃取代的铜基光敏剂CuPS 1~CuPS 6,它们在均相光解水体系表现出一定的光敏活性,1‑萘基取代的CuPS 3有最大的制氢活性,制氢TON最高可达957。通过对氮配体(NN 2~NN 6)的理论计算对比分析,NN 3的1‑萘基取代基与菲咯啉母核具有较大的二面角,对铜配合物的激发态有较好的保护作用,有利于提高制氢效率。蒽基和芘基由于其自身π‑π堆积,不利于电子传输,因此降低了CuPS 5和CuPS 6的制氢活性。荧光猝灭实验表明,氧化猝灭为主要猝灭途径。
猜你喜欢
化工管理(2022年14期)2022-12-02
高等学校化学学报(2022年10期)2022-10-14
化工学报(2021年1期)2021-01-30
中国特种设备安全(2020年11期)2020-06-09
上海建材(2020年12期)2020-04-13
山东化工(2019年2期)2019-02-16
通信电源技术(2018年5期)2018-08-23
浙江大学学报(工学版)(2016年2期)2016-06-05
印制电路信息(2015年3期)2015-02-05
