
水液相环境下α-丙氨酸二价锌配合物的手性转变机理*
2021-09-10 02:33:22苏丹孙玉锋郝成欣姜春旭张雪娇丛建民王佐成
中山大学学报(自然科学版)(中英文) 2021年4期
苏丹,孙玉锋,郝成欣,姜春旭,张雪娇,丛建民,王佐成
1. 白城医学高等专科学校药学系,吉林白城 137000
2. 白城市传染病医院质量控制科,吉林白城 137000
3. 白城师范学院理论计算中心,吉林白城 137000
4. 白城师范学院生命科学学院,吉林白城 137000
过渡金属锌是生命的重要微量元素,在生命体内以二价锌离子(Zn2+)形式存在,参与蛋白质的代谢过程,对骨骼和皮肤的发育及消化和代谢具有重要作用。其氨基酸配合物参与生命体内的多种代谢及生理生化过程,又可用于治疗疾病,与传统无机盐相比,具有生物学价值高、利于吸收、污染少等优点,还具备某些特别的生理和药理作用[1-3]。
α-丙氨酸(α-alanine,α-Ala)是结构最简单的手性氨基酸,是蛋白质和酶的基本单元,是生物体内的重要配体。根据旋光性可分为左-丙氨酸(L-α-Ala)和右-丙氨酸(D-α-Ala),根据构型可分为S-α-丙氨酸(S-Ala)和R-丙氨酸(R-α-Ala)。旋光性不同的α-Ala 具有不同的生理作用,生命体内L-α-Ala 有活性,D-α-Ala 过量会导致某些疾病或衰老[4-6]。金属离子α-Ala 配合物也具有手性,不同旋光性的手性分子对生命体具有迥然不同的作用,某种手性对生命体具有积极的作用,而其手性对映体则无用甚至有害。因此,光学纯的手性氨基酸及其金属配合物是否容易消旋对生命的健康极为重要。
基于手性氨基酸及其金属配合物消旋反应的重要性,人们对氨基酸及其金属配合物的手性转变做了大量的研究。文献[7-12]研究表明,α-Ala 在气相不能消旋,在水汽环境下可微量消旋,在水液相下可少量地消旋,酸碱性环境能使消旋速度大幅增加。文献[13]的研究发现,丝氨酸的旋光异构可以在水分子簇的催化与水溶剂的助催化作用下实现,羟自由基的存在可以加速丝氨酸的旋光异构,也可以导致丝氨酸损伤。文献[14]研究表明,SWBNNT(5,5)对Ala 的手性转变反应具有明显的限域催化作用。文献[15]研究表明,羟自由基水分子簇与MOR 分子筛的共催化可使Ala 极缓慢地消旋。文献[16-19]的研究表明,气相环境下α-Ala 的Cu2+、K+、Ca2+及Zn2+配合物的消旋反应难以实现。
目前,关于氨基酸锌的研究仅限于锌离子及水分子的配位对甘氨酸和两性离子甘氨酸结构的影响、锌离子对甘氨酸的配位能力、甘氨酸锌分子内的H 迁移以及气相环境下α-丙氨酸锌的消旋反应[19-22],对水液相下α-丙氨酸锌旋光异构的研究鲜见报道。为揭示α-丙氨酸锌给生命体补锌及丙氨酸是否安全,本工作采用密度泛函理论研究了水液相下丙氨酸锌的手性转变。
1 研究与计算方法
采用对处理含过渡金属和弱键作用体系具有较高精度的杂化泛函M06 方法[23-24],结合自洽反应场理论的SMD 模型[25]方法。在6-31++G(d,p)基组下优化反应过程中单重态(计算表明本工作研究的体系在可能的自旋态1、3、5、7 中,单重态最稳定)势能面上的驻点结构,同时获得吉布斯自由能热校正;采用自然键轨道NBO(natural bond orbital)方法计算了相关体系的NPA(natural population analysis)电荷;通过对过渡态[26]进行IRC(内禀反应坐标)计算[27],确认过渡态的可靠性。为得到较高精度的反应过程势能面,采用描述过渡金属有较好表现的MN15[28]泛函,在6-311++G(3df,2pd)高角动量基组下计算单点能。总自由能用Gtotal=Gtc+ESP计算(Gtc和ESP分别是自由能热校正和单点能,自由能校正温度是298.15 K)。水液相下S-型Ala与Zn2+的配合物Ala·Zn2+@W记作S-A@W,S-A@W 在a 通道异构的第1 个S-型过渡态记作S-T1@Wa,第1 个S-型中间体记作SI1@Wa;a 和b 通道共用的结构X 记作Xa(b)@W;在a 通道5 个H2O 分子 与S-A 的Zn 配位,同 时2 个 水分子簇与S-A 氢键作用的体系,记作S-A_1←5H2O·(H2O)2@Wa,其他体系表示法相似。文中计算工作采用Gaussian16[29]程序。
2 结果与讨论
把文献[19]的两种稳定构型的Ala·Zn2+配合物在水液相下优化,得到S-A_1@W 和S-A_2@W及其手性对映体见图1。

图1 水液相下两种丙氨酸二价锌配合物的几何构型Fig.1 Geometric configuration of two alanine divalent zinc complexes in water/liquid phase
计算表明,S-A_1@W 相对于S-A_2@W 的吉布斯自由能是-22.7 kJ·mol-1,S-A_1@W 比S-A_2@W 的稳定性好。文献[19]研究表明,S-A_2@W 经过10O—9C 内旋转和螯合环打开的过渡态向S-A_1@W 异构,然后再向R-A_1@W 异构,是其手性转变的优势通道。因此为节省篇幅,本工作只讨论S-A_1@W 的手性转变(在下文将S-A_1@W简单的标记为S-A@W)。
文献[19]研究表明,S-A 可通过以氧为桥、氮为桥、锌为桥、氧与氮联合为桥以及氧与甲基碳联合为桥等多种途径进行氢迁移,实现手性转变。其中只以氧为桥的通道为第2优势通道;螯合环打开与质子从质子化氨基向羰基氧迁移协同完成后,α-氢分别以锌和氮为桥迁移的通道为第1优势通道和第3优势通道。其他通道无法通过水分子做H 迁移桥梁降低决速步能垒变为优势通道,因此为节省篇幅,水溶剂环境下S-A 的手性转变只讨论以氧、锌和氮为H 迁移桥梁的3个通道,分别命名为a、b和c通道。在水液相下,对于非H迁移反应,只需考虑隐性水溶剂效应,显性水溶剂效应可以忽略;对于H 迁移反应,除了要考虑隐性水溶剂效应,通常还要考虑显性水溶剂效应,即水分子(簇)做H 迁移媒介的情况。下面对a、b和c 通道分别进行讨论,a、b 和c 通道的反应历程分别见图2、图3 和图4,反应过程的势能剖面分别见图5A、图5B的b线、图5B和图5C的c线。
2.1 水溶剂环境下S-A在a通道的手性转变
该通道的2 个基元反应都是水分子(簇)做H迁移媒介的情况。Zn配位数的最大值是6,反应物S-A 的14Zn 已与2 个O 配位,考虑到水分子的作用,14Zn还要与4个水分子配位。计算表明,对于本体系单个水分子及3个水分子簇做媒介时H 迁移反应能垒高于2水分子簇做媒介,为节省篇幅,本小节只讨论2个水分子簇做媒介的H 迁移反应(单个水分子及3个水分子簇做媒介时,过渡态构象相对不稳定)。因此我们研究2 个水分子簇在S-A 的前面与1C 和10O 氢键作用,形成的反应物配合物复合物S-A←4H2O·(H2O)2m@Wa(m 表示2 聚水在S-A的前面)的异构。反应历程及过渡态矢量见图2,反应的吉布斯自由能势能剖面见图5A。
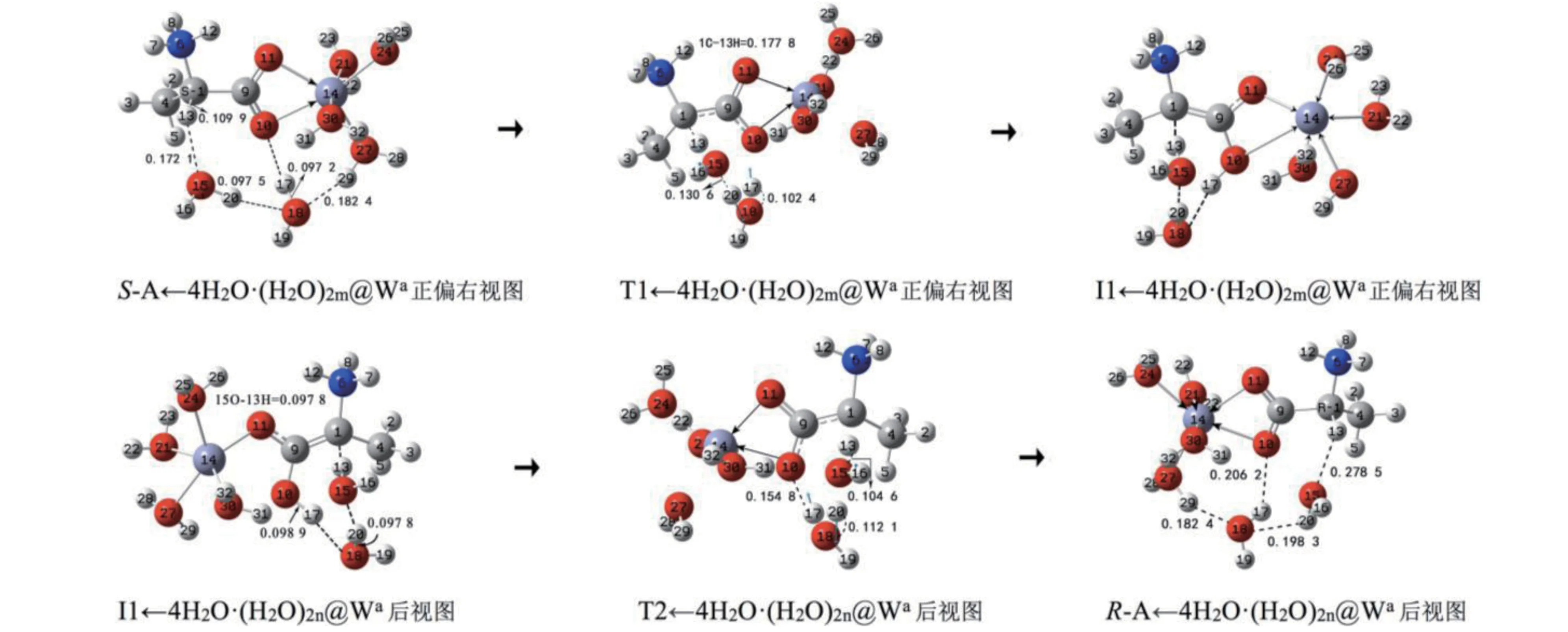
图2 水液相下S-A在a通道的手性转变历程(键长单位:nm)Fig.2 The chiral transition process of S-A in pathway a in water/liquid phase(Bond length unit:nm)
第1基元反应,是S-A的α-氢13H在水分子簇作用下从1C 向羰基氧10O 迁移。而后S-A←4H2O·(H2O)2m@Wa经过渡态T1←4H2O·(H2O)2m@Wa,异构成中 间 体I1←4H2O· (H2O)2m@Wa。 从S-A←4H2O·(H2O)2m@Wa到T1←4H2O· (H2O)2m@Wa过 程, 1C—13H、 15O—20H 和 10O—17H 键 从 0.109 9、0.097 5 和0.097 2 nm 分 别 拉 伸 至0.177 8、0.130 6和0.102 4 nm 断裂,骨架二面角6N—1C—4C—9C 从121.5°变 为133.4°,15O 和31H 以 及18O 和29H 之间的氢键断裂。多个化学键的拉伸断裂、骨架的形变以及氢键的断裂需要一定的能量。由于过渡态的氢键角1C—13H—15O、15O—20H—18O 和18O—17H—10O 分别是159.0°、159.9°和155.7°,接近平角,氢键较强,导致过渡态相对较稳定,但结构分析表明过渡态八元环结构偏离平面,又导致过渡态不稳定。因此T1←4H2O·(H2O)2m@Wa产生的内禀能垒不很高又不很低,能垒值是159.9 kJ·mol-1。与2.1 节中气相的此基元反应过渡态T1_1a产生的内禀能垒305.9 kJ·mol-1相比较,水分子(簇)及水溶剂效应的共同作用使该基元的内禀能垒大幅降低。
第2 基元反应,为使问题简化,中间体I1←4H2O·(H2O)2m@Wa的继续异构,考虑其(H2O)2m与体系的氢键解离,(H2O)2在纸面里与I1←4H2O@Wa通过氢键作用形成与I1←4H2O·(H2O)2m@Wa镜像对称的中间体反应物I1←4H2O·(H2O)2n@Wa( n 表示2 聚水在S-A 的前面) 。I1←4H2O·(H2O)2n@Wa向R-型产物的异构与第1 基元反应镜像对称,I1←4H2O·(H2O)2n@Wa经过渡态T2←4H2O·(H2O)2n@Wa,异构得到产物配合物复合物R-A←4H2O·(H2O)2@Wa,S-A@W 实现了手性对映体转变。从I1←4H2O·(H2O)2n@Wa到T2←4H2O·(H2O)2n@Wa过 程,15O—13H、 18O—20H 和 10O—17H 键 从 0.097 8、0.097 8 和0.098 9 nm 分 别 拉 伸 至0.104 6、0.112 1 和0.154 8 nm 断裂,3 个O—H 键幅度不大的拉伸所需能量不是很多,小于3 个C—H 键断裂所需能量,并且过渡态的八元环存在3条较强的氢键,导致过渡态较稳定,因此T2←4H2O·(H2O)2n@Wa产生的内禀能垒不高,只有54.2 kJ·mol-1。
2.2 水溶剂环境下S-A在b和b*通道的手性转变
文献[12]的研究表明,水分子(簇)做H 迁移媒介会使氨基和羧基间的H 迁移能垒升高,因此对于氨基和羧基间的H 迁移不讨论水分子的作用。计算表明,水分子存在时水分子的O 优先与S-I2b的14Zn 配位,水分子(簇)无法在α-C 和14Zn之间传递H,因此,对于α-C 和14Zn 之间的H 迁移不讨论显性水溶剂效应。水液相下S-A 在b通道的手性转变历程见图3,反应势能面见图5B 的b线。水液相下从S-I1@Wb还可以直接实现13H 向14Zn 迁移,命名为b*分通道,反应势能面见图5B的b*线。
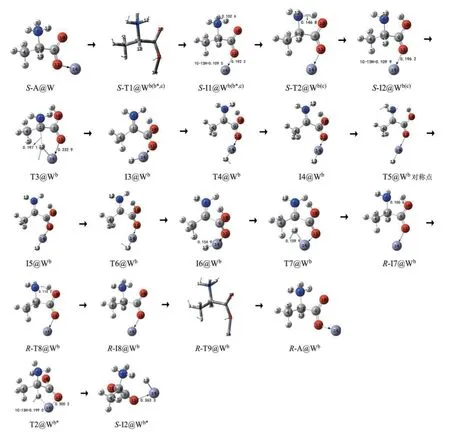
图3 水液相下S-A在b通道的手性转变历程(键长单位:nm)Fig.3 The chiral transition process of S-A in pathway b in water/liquid phase(Bond length unit:nm)
第1 基元反应(b 和b*共用),S-A@W 经11O—9C 内旋转的过渡态S-T1@W(b,b*),14Zn从去质子羧 基 内 侧 转 到 外 侧,异 构 成S-I1@W(b,b*),ST1@W(b,b*)。S-T1@W(b,b*)产 生 的 内 禀 能 垒 是24.2 kJ·mol-1。
第2 基 元 反 应,S-I1@W(b,b*)经8H 从 质 子 化氨基向羰基氧10O 迁移的过渡态S-T2@Wb,异构成S-I2@Wb。S-T2@Wb产生的内禀能垒是40.4 kJ·mol-1。
第3 基元反应,S-I2@Wb经13H 从1C 向14Zn迁移的过渡态T3@Wb,异构成I3@Wb。从S-I2@Wb到T3@Wb,1C—13H 键 长 从0.109 9 nm 拉 伸 至0.197 1 nm 断裂,11O—14Zn 键长从0.196 2 nm 拉伸 至0.232 9 nm;二 面 角6N—1C—4C—9C 从119.2°变为153.6°,骨架形变明显;二面角6N—1C—9C—10O 从-6.8°变为-59.5°,9C—1C 键右视顺时针内旋转。因此从S-I2@Wb越过T3@Wb需很多能量,T3@Wb产生了177.2 kJ·mol-1的内禀能垒。该能垒远高于气相下此基元的内禀能垒1.1 kJ·mol-1[19],说明水溶剂对此基元反应起了极显著的负催化作用。原因有两个,一是S-I2@Wb与S-I2b相比较,1C 的电荷从-0.025 e 增加到-0.183 e,13H 的电荷从0.007 e 增加到0.286 e,1C—13H 键长从0.127 0 nm 缩短到0.109 9 nm,1C 对13H 的库仑引力显著增加;振动频率分析表明,1C—13H键的伸缩振动频率从1 281.6 cm-1增加到3 062.3 cm-1,发生了极显著的蓝移。二是从S-I2 @ Wb到T3_1@Wb,虽然13H 的电荷从0.286 e 逐渐变为-0.51 e,但1C 的电荷从-0.183 e 逐渐变为0.341 e,对13H负离子具有库仑引力作用。
第4基元反应,I3@Wb经13H 相对于14Zn左右翻转的过渡态T4@Wb,异构成I4@Wb。T4@Wb产生的内禀能垒很小,只有2.4 kJ·mol-1。
第5 基元反应,I4@Wb经6N—1C 和9C—1C 键协同内旋转的过渡态T5@Wb,异构成与I4@Wb镜像对称的中间体配合物I5@Wb。从I4@Wb到T5@Wb过程,9C—1C键右视逆时针转内旋转45.0°,还要克服来自12H 的很大的空间位阻,同时6N—1C 键俯视顺时针内旋转127.0°,需要很高的能量,因此T5@Wb产生了197.2 kJ·mol-1的内禀能垒。
第6基元反应,I5@Wb经13H 相对于14Zn左右翻转的过渡态T6@Wb,异构成I6@Wb。T6@Wb产生的内禀能垒很小很小,只有1.6 kJ·mol-1。
第7 基元反应,I6@Wb经过渡态T7@Wb,13H 从14Zn 向1C 迁移,异构成R-I7@Wb。从I6@Wb到T7@Wb,13H—14Zn 键 从0.154 9 nm 拉伸至0.159 9 nm,T7@Wb产生的内禀能垒是32.5 kJ·mol-1。
第8 基元反应,R-I7@Wb经8H 从羧基氧10O向氨基氮6N 迁移的过渡态R-T8@Wb,异构成RI8@Wb。R-T8@Wb产生的内禀能垒只有1.1 kJ·mol-1,该基元反应几乎无势垒。
第9 基元反应,R-I8@Wb经11O—9C 内旋转的过渡态R-T9@Wb,14Zn 从去质子羧基外侧转到内侧,异构成R-A@Wb,R-T9@Wb产生的内禀能垒是12.9 kJ·mol-1。结构分析表明,R-A@Wb与SA@W 镜像对称,S-A@W 在b 通道实现了手性对映体转变。
S-A@W 在b 通道的反应历程及势能面,展现了S-A@W 在b 通道手性对映体转变的过程与内在美。
b*分通道。
第2 基元,S-I1@W(b,b*)经13H 从1C 向14Zn 迁移的过渡态T2@Wb*,异构成S-I2@Wb*。T2@Wb*产生的能垒是345.6 kJ·mol-1。这远远高于T3@Wb产生的内禀能垒,原因是从S-I1@W(b,b*)到T2@Wb*,1C—13H 键长从0.109 5 nm 拉伸至0.199 0 nm,断裂, 11O—14Zn 键 长 从0.192 2 nm 拉 伸 至0.300 2 nm,断裂;二面角6N—1C—4C—9C 从119.5°变为161.7°,二面角6N—1C—9C—10O从-9.7°变为-79.3°。1C—13H键的拉伸、11O—14Zn 键的拉伸、9C—1C 键的内旋转及骨架的形变均大于从S-I2@Wb到T3@Wb过程许多。此基元能垒太高,反应通常无法实现,接下来的异构不予讨论。
2.3 水溶剂环境下S-A在c通道的手性转变
S-A 在c 通道的手性转变反应历程及过渡态见图4,反应的自由能势能面见图5B和图5C的c线。
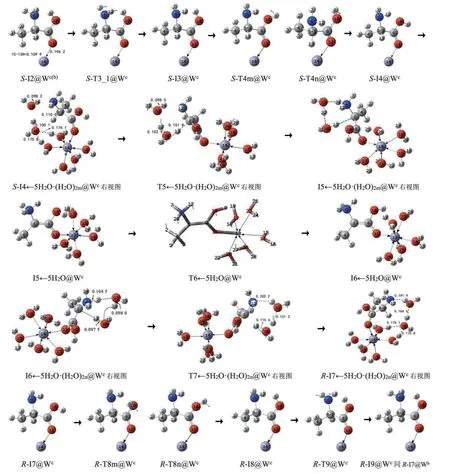
图4 水液相下S-A在c通道的手性转变历程(键长单位:nm)Fig.4 The chiral transition process of S-A in pathway c in water/liquid phase(Bond length unit:nm)
第1、2 基元反应b、c 通道公用,气相第3 基元氨基翻转与羧基旋转协同进行的过程在水液相下变成了分步进行的过程,分为第3、4 基元,原来气相的13H 从1C 向6N 迁移的第4 基元现在变为第5基元反应,是该路径的决速步。计算表明,水溶剂环境下水分子(簇)考虑与否对非H 迁移反应的能垒影响不大,对H 迁移反应的能垒影响较大。因此对第3、4 基元只考虑水溶剂效应,而对第5 基元(H 迁移反应)相似于c 通道,既考虑水分子与14Zn 配位、水分子簇作H 迁移媒介,同时又考虑水溶剂效应。为节省篇幅,突出重点,并兼顾反应的连续性,本节对第3、4 基元做一般讨论,对第5基元进行详细讨论,对后面的过程只做简单说明。
第3 基元反应,S-I2@W(b,c)经7H 与2H 左右翻转的过渡态S-T3@Wc,7H 与2H 从左侧翻转到右侧,异构成S-I3@Wc。7H 与2H 左右翻转所需能量很少,S-T3@Wc产生的内禀能垒只有15.0 kJ·mol-1。
第4 基元反应,S-I3@Wc经10O—9C 键内旋转的过渡态S-T4m@Wc(或S-T4n@Wc),羧羟基俯视逆时针转(或顺时针)旋转,8H 从羧基外侧转到内侧,异构成S-I4@Wc。化学键内旋转所需能量不多,S-T4m@Wc和S-T4n@Wc产生的内禀能垒分别是48.6和48.9 kJ·mol-1。
第5 基元反应,是S-I4@Wc的α-氢在水分子簇作用下从1C向6N迁移。首先14Zn与5个水分子配位(Zn 配位数的最大值是6,14Zn 已与一个O 配位),2 个水分子簇与6N、13H、28H 以及23H 氢键作用,形成中间体反应物S-I4←5H2O·(H2O)2m@Wc。而后S-I4←5H2O·(H2O)2m@Wc经过渡态T5←5H2O·(H2O)2m@Wc,异构成中间体I5←5H2O·(H2O)2m@Wc。 从S-I4←5H2O· (H2O)2m@Wc到T5←5H2O·(H2O)2m@Wc,1C—13H 键 从0.110 0 nm 拉 伸 至0.151 8 nm,断裂,30O—32H 键从0.100 1 nm 拉伸至0.102 9 nm,断裂,33O—34H键从0.098 2 nm拉伸至0.098 5 nm;二面角6N—1C—4C—9C 从127.5°变为157.5°;二面角14Zn—11O—9C—10O从178.3°变为-65.1°,14Zn←5H2O 集团向纸面里大幅翻转;30O—23H 及30O—28H 间的氢键断裂。多个共价键和氢键断裂、骨架形变、及14Zn←5H2O集团的大幅翻转需一定的能量,T5←5H2O·(H2O)2m@Wc产 生 的 内 禀 能 垒 较 高, 是140.7 kJ·mol-1。
第6 基元是羧羟基H 和Zn 在纸面里外翻转过程。羧羟基H 和Zn 在纸面里外翻的能垒与水分子簇是否与质子化氨基上的H 有氢键作用影响甚微,为使问题简便,此基元反应物不考虑I5←5H2O·(H2O)2m@Wc的(H2O)2m。但由于配位键较强,且Zn在纸面里外翻转所需能量的多少与是否有水分子与其配位密切相关,此基元仍考虑有5个水分子与I5 配位。因此,把此基元的反应物视为I5←5H2O@Wc。I5←5H2O·(H2O)2m@Wc经10O—9C 和11O—9C 协同内旋转的过渡态T6←5H2O@Wc,8H从纸面外转到纸面里,Zn与5个水集团从纸面里转到纸面外,异构成I6←5H2O@Wc。此基元是2 个化学键内旋转,所需能量很少,T6←5H2O@Wc产生的内禀能垒大约是6.5 kJ·mol-1。
第7 基元反应与本小节第5 基元反应镜像对称。I6←5H2O@Wc的质子化氨基的7H 与纸面里的2 个水分子簇氢键作用,形成此基元的反应物I6←5H2O·(H2O)2n@Wc。I6←5H2O·(H2O)2n@Wc经3 质子协同迁移的过渡态T7←5H2O·(H2O)2n@Wc,异构成中间体产物R-I7←5H2O·(H2O)2n@Wc,S-A@W 实现了手性转变。从I6←5H2O·(H2O)2n@Wc到T7←5H2O·(H2O)2n@Wc,6N—7H、33O—32H 和30O—13H 键拉伸断裂所需能量不是很多,且T7←5H2O·(H2O)2n@Wc的七元环结构存在较强的氢键,构象相对稳定,因此T7←5H2O·(H2O)2n@Wc产生的内禀能垒不高,是83.2 kJ·mol-1。
第8 基元反应与本小节第4 基元反应镜像对称,羧羟基H 的旋转能垒与Zn 是否与水分子配位关系不大,为使问题简便,将R-I7@Wc视为此基元反应物。R-I7@Wc经10O—9C 键内旋转的过渡态R-T8m@Wc或R-T8n@Wc,8H 在纸面外(或纸面内)从羧基内侧转到外侧,异构成R-I8@Wc。R-T8m@Wc和R-T8n@Wc产生的内禀能垒分别是46.1和45.9 kJ·mol-1。
第9基元反应与本小节第3基元反应镜像对称。R-I8@Wc经氨基氢左右翻转的过渡态R-T9@Wc,7H 和12H 从6N 的右侧翻转到左侧,异构成RI9@Wc。R-T9@Wc产生的内禀能垒是4.0 kJ·mol-1。
R-I9@Wc同于R-I7@Wb,其接下来的第10 和11基元,同于R-I7@Wb的异构,见2.1.2小节S-A在b 通道手性转变的第8 和9 基元。最终得到产物R-A@Wc,S-A@W在c通道实现手性对映体转变。
S-A@W 在c 通道的反应历程及势能面,同样展现了手性对映体转变的过程美与内在美。
从图5 可以看出,S-A@W 手性转变的优势通道是c,第5 基元是决速步骤,能垒是140.7 kJ·mol-1。亚优势通道是a,决速步是第1 基元,能垒是159.9 kJ·mol-1。气相反应的优势通道b 在水液相下变为劣势通道,第5 基元是决速步,能垒是194.2 kJ·mol-1。从前面的讨论可知,此能垒来自α-H 从α-C 向Zn 迁移的过渡态,然而气相下该通道的决速步能垒来自第2 基元9C—1C 键内旋转的过渡态[19]。溶剂效应改变了反应通道优劣的排序及决速步骤。140.7 kJ·mol-1远远大于温和反应能垒80.0 kJ·mol-1[30],距离化学反应极限能垒167.0 kJ·mol-1[30]不算太远,说明通常水液相下S-A 只能微量地消旋。
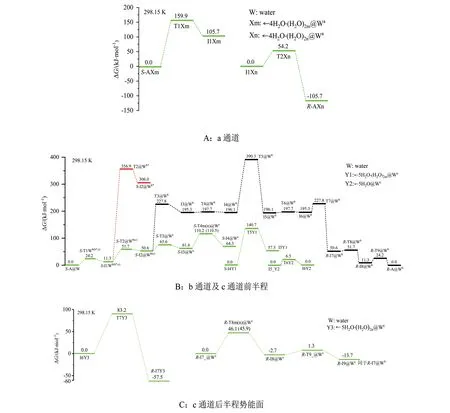
图5 水液相下S-A手性转变反应的吉布斯自由能势能面Fig.5 Gibbs free energy potential energy surface of S-A chiral transition reaction in water/liquid phase
3 结 论
在SMD/MN15/6-311++G (3df, 2pd)//SMD/M06/6-31++G(d, p)双水平,对两性丙氨酸二价锌配合物S-A 在水液相下手性转变的研究得到如下结论:
1)S-A@W 在c 通道的手性转变反应最具优势,决速步自由能垒是140.7 kJ·mol-1,来自α-H从α-C向氨基N迁移的过渡态。
2)S-A@W 在a 通道的手性转变反应是亚优势通道,决速步自由能垒是159.9 kJ·mol-1,来自α-H从α-C向羰基O迁移的过渡态。
3)S-A@W 在b 通道的手性转变反应为劣势通道,决速步自由能垒是194.2 kJ·mol-1,来自α-H从α-C向Zn迁移的过渡态。
结果表明,丙氨酸锌在水液相下较难消旋,其用于生命体补锌及丙氨酸具有比较好的安全性。
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
中华养生保健(2020年3期)2020-11-16 00:52:28
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
中外医疗(2015年11期)2016-01-04 03:58:55
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
