
病毒调节宿主适应性免疫的分子机制
2021-08-31 01:21徐乐乐李志伟马志倩肖书奇
中国动物传染病学报 2021年4期
徐乐乐,李 洋,李志伟,马志倩,肖书奇
(西北农林科技大学动物医学院,杨凌 712100)
免疫系统是一个复杂的网络,高等哺乳动物的免疫系统主要分为先天性免疫系统和适应性免疫系统,它们共同保护机体免受病原体的侵害。
先天性免疫系统是宿主防御病毒感染的第一道防线,由先天免疫细胞群(如髓样细胞、NK细胞和先天淋巴样细胞),以及体液免疫系统(如防御素和补体)等组成[1]。先天免疫系统利用进化保守的模式识别受体系统,如Toll样受体(toll like receptor,TLRs)、C型凝集素受体(C-type lectin receptors,CLR)、NOD样受体(NOD like receptor,NLRs)和RIG-1样受体(RIG-1 like receptor,RLRs)等[2]识别病毒。
在先天免疫系统无法应对病毒感染时,机体就需要调动适应性免疫系统来清除病毒。适应性免疫系统主要是通过T细胞和B细胞特异性识别入侵的病毒,并将其清除。病毒感染后诱发机体产生病毒特异性CD4+T细胞和CD8+T细胞。幼稚CD4+T细胞在接受抗原提呈细胞(antigen-presenting cell,APC)提呈的MHCⅡ(major histocompatibility complex Ⅱ)识别的表位后,在不同细胞因子环境下分化成不同类型的辅助性T细胞(T helper cells,Th),Th细胞通过分泌细胞因子等来调控其他免疫细胞,也可作用于B细胞而使B细胞分化成浆细胞。CD8+T细胞,又称为细胞毒性T细胞(cytotoxic T lymphocyte,CTL),它可直接与被感染的靶细胞特异性地结合而杀伤靶细胞。B细胞主要参与体液免疫,机体可以通过效应B细胞产生特异性抗体中和病毒[1,3]。但对于有些病毒,在存在非中和或亚中和浓度的抗体时,抗体不但不能抑制病毒感染,反而会产生抗体依赖增强现象(antibody dependent enhancement,ADE),促进病毒的感染。
一般认为NK细胞是先天免疫系统的组成部分,但研究发现NK细胞也具有适应性免疫系统的特征,例如病原体特异性的扩增,持续存在的“记忆”细胞。NK细胞的这一特性,使其成为改进疫苗、开发新型免疫疗法的重要靶细胞。
在宿主和病毒的长期斗争中,病毒进化了各种免疫逃逸机制以逃避宿主的免疫应答。病毒可以通过自身的突变、糖基化等方式,遮盖有效的中和表位,干扰中和抗体的产生与识别。同时病毒也会干扰宿主的细胞免疫反应,帮助病毒逃避免疫反应的清除。
1 病毒调节T细胞介导的免疫应答
T细胞在抗原特异性免疫反应的发展和调节中起着核心作用,包括直接杀伤靶细胞,激活B细胞,调节抗原提呈环境中的细胞因子环境,并监控和调节免疫反应以控制炎症反应等。
登革热病毒(Dengue virus,DENV)感染过程中T细胞起到重要保护作用。原发性感染1型DENV导致1型DENV特异性CD4+和CD8+T细胞的激活。激活的CD4+T细胞为B细胞提供帮助。产生的1型DENV特异性抗体,可有效清除DENV感染。1型DENV特异性记忆CD4+和CD8+T细胞保留在T细胞库中,二次同源攻击会唤起记忆反应,识别相同的MHC-肽,从而有效的控制感染[4]。
急性DENV感染期间,滤泡辅助T细胞(follicular helper T cell,Tfh)增多,一般来说,Tfh细胞能够在生发中心之间流动,促进Th的多样化。DENV感染中,Tfh细胞可促进DENV特异性抗体成熟,介导更高的中和抗体能力[5]。DENV感染激活效应CD4+T细胞也可有细胞毒性功能,这些可能是具有效应记忆表型(CD45RAhiCCR7low)的T效应记忆RA细胞(TEMRA细胞)。DENV特异性CD4+TEMRA细胞表达粘附蛋白G蛋白偶联表面受体56(GPR56)、趋化因子受体CX3CR1和丝氨酸蛋白酶颗粒酶,是IFN-γ的重要来源[6-7]。但更多时候,病毒不会按设想的方式激活T细胞免疫反应。对于急性原发性DENV感染的患者,CD8+T细胞反应在退热前后达到峰值。在疾病的发热期,活化的CD8+T(CD38+HLA-DR+)细胞大量扩张。然而,在体外分析时,发现这些细胞活性已耗尽,对聚集的DENV肽刺激无反应,产生IFN-γ和IL-2的能力有限,这种衰竭可能缘于T细胞受体(T-cell receptor,TCR)不足[4,8]。
猪无法对猪繁殖与呼吸综合征病毒(Porcine reproductive and respiratory syndrome virus,PRRSV)产生有效免疫的主要原因之一可能是T细胞反应不足。在感染PRRSV的第一周,CD4+/CD8+T细胞比例出现较大的瞬时下降。该变化可能是由于凋亡引起CD4+T细胞的暂时丢失或抗原特异性CD8+T增殖引起的[9]。
接种PRRSV疫苗35 d后,可观察到低病毒特异性IFN-γ分泌的CD8+T细胞反应[10]。感染前CD8+T细胞的暂时耗竭并没有导致感染的增加,这表明CTL在控制急性感染中没有功能性作用[11-12]。感染后第14 d观察到记忆CTL增殖,但直到感染后第49 d才检测到CTL活性[13]。表达CD8的T细胞细胞毒效应功能与PRRSV感染的应答关系仍需更多探究。
Th17细胞可以通过产生促炎性细胞因子,如IL-17A、IL-17F和IL-22在胞外细菌免疫中发挥重要作用[14]。高致病性PRRSV株(HP-PRRSV)病毒可特异性抑制猪外周血和肺中的Th17细胞的功能,导致猪对继发性细菌感染的敏感性增加[15]。
2 病毒调节B细胞介导的免疫应答
通常抗原可被B细胞受体(B-cell receptor,BCR)识别、内吞、降解,然后通过MHCⅡ提呈到细胞表面,寻找对同一抗原特异性分化的CD4+T细胞。T细胞识别出MHCⅡ提呈的抗原后,通过分泌细胞因子刺激B细胞增殖并分化,一些细胞分化成分泌免疫球蛋白IgM的浆细胞,其他细胞迁移到B细胞滤泡中,在CD4+Tfh和滤泡树突状细胞分泌的细胞因子的帮助下,形成生发中心。在生发中心,B细胞增殖并经历体细胞超突变和同种型转换。浆细胞产生的亲和力成熟的抗体和在抗原识别后增强抗体效价的记忆细胞,两者构成了抵御感染的重要防线[16-17]。多数情况下,抗体与病毒结合后可阻止病毒附着或进入靶细胞,之后通过激活补体、抗体依赖细胞介导的细胞毒性作用(antibody-dependent cell cytotoxicity,ADCC)等方式清除病毒。
感染DENV 4~7 d,IgG含量已达全部DENV特异性抗体的50%。DENV感染可以提高血液和淋巴结中的CD14+CD16+细胞数,CD14+CD16+细胞促进B细胞分化成浆细胞,增强IgG和IgM的分泌[4,18]。在继发性DENV感染期间,B细胞和T细胞激活反应的幅度和动力学明显增强,这些记忆B细胞具有DENV特异性,可快速分泌大量病毒特异性IgG,且浆细胞显著增多,可达正常水平的1000倍[19]。免疫过或感染过DENV的患者更有可能发展登革热出血热(dengue hemorrhagic fever,DHF)/登革热休克综合征(dengue fever shock syndrome,DSS)的原因可能是ADE。表达Fc受体(Fc receptor,FcR)的细胞,如树突状细胞(dendritic cells,DC)和单核细胞,可能发生ADE,导致高水平的病毒复制。抗体也会增加病毒感染细胞的感染范围,使本身不易感的细胞感染。这种抗体介导的ADE效应,本文将在后文中另作阐述。
病毒在很多时候不能被预期的B细胞免疫反应清除。在PRRSV感染的早期,可以检测到强烈的PRRSV抗体反应,但早期产生的抗体不能中和PRRSV,具有PRRSV中和活性的血清抗体仅在感染后较晚的时间出现。接种疫苗或先前感染的动物在异源攻击后可以观察到记忆性中和抗体,但记忆性中和抗体反应需要两次感染间隔2~3个月,若再次感染间隔1个月或更少,则检测不到记忆中和抗体反应[14]。
在持续感染期间,通常会观察到抗体产生的明显滞后,甚至无法产生中和抗体,这很可能导致某些病毒无法被清除。在淋巴细胞性脉络膜脑膜炎病毒(lymphocytic choriomeningitis virus,LCMV)模型中观察到,LCMV特异性B细胞在持续感染的几天内迅速衰竭,阻断Ⅰ型干扰素(IFN-Ⅰ)介导的信号传导可完全逆转这种衰竭。在早期干扰IFN-Ⅰ信号促进了LCMV特异性B细胞的存活和分化,从而加速了中和抗体的产生。这种改善不依赖于B细胞中IFN-Ⅰ信号的停止,而是依赖于病毒特异性CD8+T细胞反应的改变。CTL在感染的前几天以穿孔素依赖性方式有效参与并杀死了LCMV特异性B细胞。通过促进CTL功能障碍来阻断IFN-Ⅰ信号可保护LCMV特异性B细胞[20]。
人类免疫缺陷病毒(Human immunodeficiency virus,HIV)感染会诱导免疫球蛋白G3(immunoglobulin G3,IgG3),IgG3可以阻止免疫系统中的B细胞完成正常的抗病毒功能。这种现象可能是机体试图减少因HIV引起的免疫系统过度活跃而导致的潜在破坏性影响,但这样做也会损害正常的免疫功能。IgG3通过结合B细胞受体,阻止它对病毒或其他目标的反应。通过治疗,慢性感染者体内病毒得到控制时,IgG3会停止与B细胞受体结合,说明IgG3的活性与慢性感染期间HIV的存在直接相关[21]。
3 NK细胞与适应性免疫
3.1 病毒介导的具有适应性免疫特点的NK细胞免疫反应传统上认为天然杀伤(natural killer,NK)细胞不具有记忆性,是先天免疫系统的主要效应细胞,可以迅速消除肿瘤和病毒感染的细胞。但研究发现,NK细胞的功能远比当前所认识的要多样化。鼠和灵长类NK细胞的亚群能够进行抗原依赖性扩增和长时间的免疫记忆。NK细胞功能可能兼顾先天和适应性免疫[22]。
人类巨细胞病毒(Humancytomgalo virus,HCMV)感染可刺激表达NKG2C的记忆性NK细胞频率升高和变化。移植经HCMV活化的造血细胞的患者中,NKG2C+NK细胞数量也会增加并长期持续存在[23-25]。NKG2C+NK细胞还可以抗体依赖的方式对HCMV感染的细胞表现出增强的功能性反应。首次与抗体结合后,这些细胞被表观遗传修饰且更长寿,并能够在新的抗体结合后显著增强抗体依赖性功能[26]。
小鼠继发性感染流感病毒后,可以激活记忆NK细胞,并产生保护性反应[27-28]。对小鼠进行流感疫苗接种也可促进记忆性NK细胞的发育,这些细胞在受到病毒攻击后会迅速分泌大量IFN-γ[29]。NK细胞可对流感病毒、水疱性口炎病毒(Vesicular stomatitis virus,VSV)或HIV的疫苗产生特异性记忆[30]。这些研究可以为理解流感病毒和其他病毒如何靶向NK细胞,产生记忆NK细胞提供重要参考。
研究发现了NK细胞的一个独特亚群,其特征是FcRγ表达不足[26,31]。这些FcRγ缺乏NK细胞(FcRγ-NK),也称作“g-NK cells”。存在HCMV特异性抗体的情况下,FcRγ-NK对HCMV感染细胞的功能反应大大增强,优于常规NK细胞[31],这表明FcRγ-NK细胞对HCMV感染的细胞可以不通过CD16介导的抗体依赖性识别。
3.2 NK细胞记忆产生的分子机制CXCR6是肝脏NK细胞上的趋化因子受体。NK细胞对半抗原和病毒的记忆依赖于CXCR6。CX CR6-NK细胞无法建立对半抗原或病毒的记忆反应。CXCR6抗体增强了体外记忆NK细胞的抗原特异性细胞毒性,而CXCL16减弱了这种细胞毒性[30]。
最近的研究阐述了MCMV感染时NK细胞记忆形成的分子机制。缺乏IL-12受体或STAT4信号传导的NK细胞不能克隆增殖,并且感染MCMV后生成的记忆NK细胞存在缺陷[32]。来自促炎细胞因子(包括IL-12、IL-18和IFN-Ⅰ)的信号对于驱动转录因子锌指蛋白32(Zbtb32)的表达是必要的,而转录因子Zbtb32对于MCMV感染过程中抗原特异性NK细胞的增殖和保护功能至关重要[33]。Zbtb32作为重要的检查点分子,可通过拮抗肿瘤抑制因子Blimp-1来促进活化的NK细胞的增殖。存在抗原和促炎信号的情况下,抗原特异性NK细胞的增殖也需要共刺激激活信号,感染MCMV时缺少激活受体DNAM-1的Ly49H+NK细胞无法扩增并形成长寿的记忆细胞[34-35]。因此,抗原特异性NK细胞的活化和增殖需要CXCR6、受体与抗原的结合(Ly49H-m157)、共刺激信号传导(DNAM-1)和促炎细胞因子信号传导(IL-12、STAT4、Zbtb32)。
病毒具体是如何调节NK细胞的进化,NK细胞又是如何参与适应性免疫的,仍需进一步探究。
4 病毒逃逸宿主适应性免疫
病毒有多种逃逸机制以保证自身在宿主细胞中的存活,包括逃逸抗病毒分子、逃逸抗病毒信号通路和逃逸抗病毒细胞等。病毒逃逸适应性免疫主要是通过逃逸抗病毒细胞,这包括两个方面:病毒影响T细胞发育分化和抑制T细胞功能;病毒影响B细胞发育分化以及逃逸浆细胞产生的中和抗体。
4.1 病毒逃逸T细胞介导的免疫反应病毒可以通过多种方式调节CD4+T/CD8+T细胞的活性或数量,干扰免疫反应,从而逃避机体对病毒的清除。
PRRSV感染会诱导占有胸腺细胞总数约90%的CD4+CD8+胸腺细胞大量凋亡,使胸腺内成熟T淋巴细胞的数量减少,导致外周T淋巴细胞对抗原的识别及细胞免疫相关细胞因子分泌异常。外周血中与中和抗体产生相关的Tfh数量的改变,会导致淋巴结生发中心中可产生高亲和力中和抗体的B细胞选择失败,使感染PRRSV的猪中和抗体产生推迟[36]。
PRRSV感染造成胸腺CD4+CD8+凋亡原因可能有以下几种解释。PRRSV感染期间会造成骨髓细胞凋亡,使从骨髓迁移到胸腺的胸腺细胞数量不足,加之胸腺皮质中的胸腺细胞发生凋亡最终导致胸腺萎缩[37];CD4+CD8+胸腺细胞经caspase-3、caspase-8和caspase-9激活后对糖皮质激素敏感。PRRSV-2感染可使感染猪血清糖皮质激素水平上调,并诱导胸腺内caspase-3、caspase-8和caspase-9激活[38]。抑制糖皮质激素的产生可防止病毒诱导的胸腺细胞衰竭,侧面证明PRRSV感染造成的胸腺细胞凋亡可能与血清糖皮质激素水平升高有关[36];在HIV感染中,TNF-α大量存在于巨噬细胞的膜和CD8+T细胞的TNF受体Ⅱ(TNFR2)上并造成了细胞凋亡[39]。在PRRSV-1感染的胸腺巨噬细胞的细胞质中可检测到TNF-α,胸腺髓质的中性粒细胞和淋巴细胞的细胞质中也有少量的TNF-α,推测TNF-α可能与促进胸腺细胞凋亡有关[36]。
非洲猪瘟病毒(African swine fever virus,ASFV)会影响宿主转录组,降低中性粒细胞、CD8+T效应细胞募集趋化因子,避免CD8+T效应细胞和中性粒细胞外杀菌网络(neutrophil extracellular traps,NETS )[40-41]。ASFV可下调MHCⅡ抗原加工的关键因子DMA、DMB,并上调其抑制因子DOA、DOB表达[41],干扰MHCⅠ/Ⅱ的表达,从而抑制抗原加工和提呈。ASFV的强毒株感染巨噬细胞会增加Ⅰ类MHC基因的表达,但MHCⅠ分子向质膜的传递却没有平行增加,可能原因是ASFV通过改变TGN46的定位,破坏高尔基体反面网状结构(trans-Golgi network,TGN)来降低MHCⅠ向膜表面提呈的能力或者下调蛋白酶体、溶酶体等实现对MHCⅠ/Ⅱ抗原提呈能力的调控[40,42]。对ASFV调控MHC表达差异和转运机制进一步研究,会有助于了解ASFV致病机制和逃逸机制。
甲型流感病毒(Influenza A virus,IAV)不同毒株之间的毒力不同,有的IAV感染可以在上皮细胞中逃避CD8+T细胞介导的识别和清除。存活细胞逃逸CD8+T细胞识别可能是不再表达IAV抗原,从而失去抗原加工和提呈途径,或通过细胞分裂稀释两个子细胞之间的抗原量,从而逃避CD8+T细胞的细胞溶解[43]。
抑制性配体PD-L1也会抑制CD8+T细胞反应。细胞表面PD-1的上调可导致CD8+细胞的衰竭。气管上皮细胞感染IAV后PD-L1的表达增加,对PD-L1的体内阻断可改善CD8+T细胞数量并降低病毒滴度[44],但无法改善整体疾病情况或拯救CD8+T细胞功能。诱导HBV特异性CD8+T细胞应答以及HBsAg特异性抗体产生需要CD4+T细胞。然而,在慢性HBV感染期间,HBV特异性CD4+T细胞应答也较弱[45]。可能是由于PD-1在HBV特异性CD4+T细胞上表达[46],这可能导致CD4+T细胞的免疫应答抑制。
DC是主要的抗原提呈细胞。PRRSV在DC中复制可削弱宿主针对病毒的有效免疫应答。PRRSV可能会通过调节MHCⅡ和CD80/86的表达来影响DC的基本功能。APC的损伤和/或细胞死亡不能正确激活T细胞,继而可能导致无法诱导有效免疫反应[36,45]。与PRRSV类似,HIV和丙型肝炎病毒(Hepatitis virus C,HCV)等会通过诱导Treg的增殖和活化抑制免疫反应,从而逃避免疫反应并持续感染[47-48]。PRRSV感染的DC可诱导Treg的增殖,其表型为CD4+CD8+CD25+Foxp3high[48]。Treg在体外可抑制HBV特异性免疫反应,而消耗Treg促进了HBV清除[49]。
4.2 病毒逃逸B细胞介导的免疫反应病毒可以通过变异、抗原漂移和糖基化等多种形式,直接逃逸抗体的中和作用,也可通过调节特异性CD4+T细胞、CD8+T细胞,抑制特异性B细胞分泌抗体,从而逃逸B细胞介导的免疫反应。
最初人们认为在持续性HBV(hepatitis B virus,HBV)感染期间缺乏分泌HBsAg特异性抗体的B细胞的原因,可能是HBsAg特异性CD8+T细胞对HBsAg特异性B细胞的杀伤。最近研究表明,急性或慢性HBV感染的患者之间HBsAg特异性B细胞的数量没有显著差异,这种功能障碍与CD21-CD27记忆B细胞表型有关,该表型高表达抑制性受体(包括PD-1)。清除HBsAg或PD-1可以恢复HBsAg特异性B细胞抗体产生能力[50-51]。这些数据表明,尽管在慢性HBV感染期间存在HBsAg特异性B细胞,但循环的HBsAg耗尽了其反应或使其失去了活力,造成抗原特异性免疫抑制功能。HBsAg特异性抗体应答还取决于CD4+T细胞,微弱的HBV特异性CD4+T细胞应答可能会导致HBsAg特异性B细胞衰竭。
ASFV的抗体不能有效中和ASFV的原因之一,是在传代过程中病毒的表型特征会发生改变。这种差异不是由于关键表位的抗原变异性引起的,而与ASFV病毒粒子的磷脂组成有关,这种组成随细胞培养传代的代次而不同。将磷脂酰肌醇加入高传代病毒的膜中,可使其具有与低代次病毒相似的中和敏感性。通过特异性脂肪酶从低代次病毒中去除磷脂酰肌醇,可将该病毒从可中和转变为不可中和[52-53],这表明这种膜成分对于中和抗体的表位正确展示是必不可少的。这些数据为在高代次病毒难以重复验证ASFV中和抗体提供了一种解释。
PRRSV的GP5蛋白存在诱骗表位A,在抗体产生时,优先产生针对A表位的非中和活性抗体,推迟中和抗体的产生[54]。GP5蛋白中的N-聚糖部分对于病毒逃避中和抗体很重要,诱骗表位A除了作为免疫诱饵,还可以产生额外的N-糖基化位点,进一步屏蔽中和表位B表位,推迟中和抗体的产生,并影响抗体的特异性[55]。研究还提出GP5蛋白的高变区还存在C表位,在感染后期C表位易出现突变,进一步阻断中和表位,导致病毒逃逸中和抗体和病毒血症的重新出现[56]。
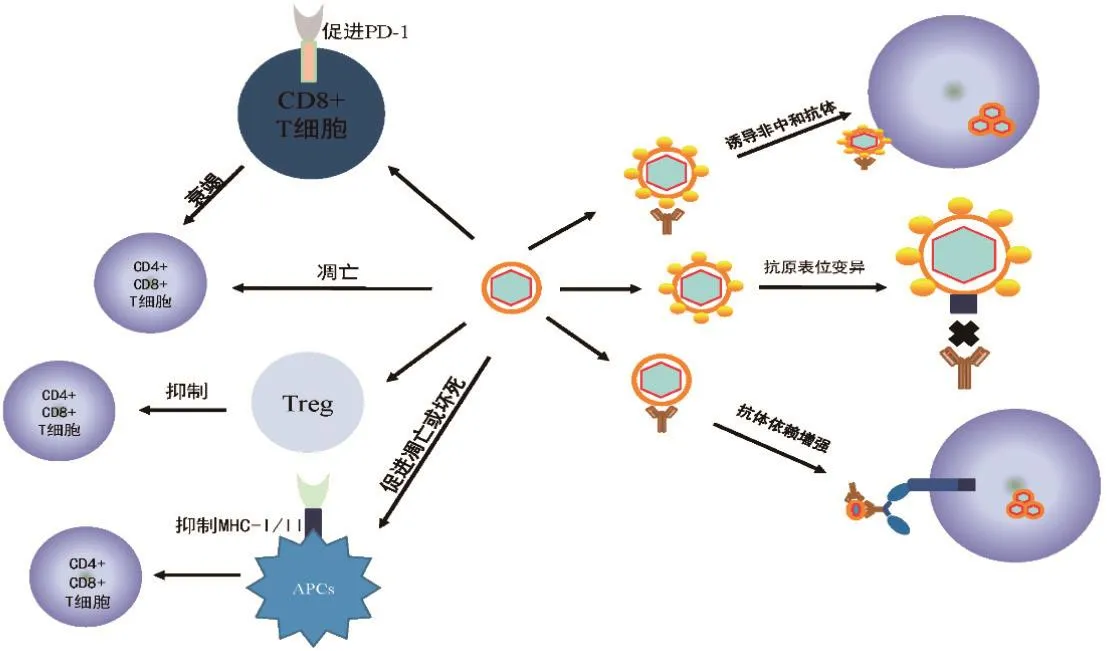
图1 病毒逃逸适应性免疫的机制Fig.1 Mechanism of virus escape from adaptive immunity
B细胞在抗原刺激下转化为浆细胞,浆细胞可通过产生抗体直接中和病毒,也可通过ADCC杀伤感染的靶细胞[56]。一些病毒在复制翻译过程中较易产生突变,如流感病毒和PRRSV等,在宿主体内复制与翻译过程中,高频率的病毒抗原表位持续变异,使病毒突变株可逃逸已有抗体的中和作用[56,58]。HIV ENV三聚体具有“开放”和“闭合”两种形态,处于“闭合”状态时,可使其对抗体有一定的抵抗力[59-60]。

表1 代表性病毒逃逸适应性免疫总结Table 1 Summary of representative virus escape adaptive immunity
5 抗体介导的抗体依赖性增强效应(antibody dependent enhancement,ADE)
病毒感染始于粘附在细胞表面,针对病毒表面蛋白的特异性抗体通常可以抑制这一过程,中和病毒,使其丧失感染细胞的能力。然而,在某些情况下,抗体在病毒感染中可起相反的作用:病毒可以“借助”抗体进入靶细胞,提高感染率,这种现象称为ADE。ADE根据其作用方式的不同,可分为内源性ADE和外源性ADE(intrinsic antibody-dependent infection enhancement,iADE)。iADE可以通过调节细胞的免疫反应,从而促进病毒感染[61-62]。抗体还可以通过诱发病毒构象改变或促进膜融合等形式诱发ADE。
抗体与病毒结合形成的抗原抗体免疫复合物与Fcγ受体(FcγR)结合后诱导iADE,触发多种免疫抑制反应促进感染。进入靶细胞的免疫复合物激活负性调节因子二羟丙酮激酶(dihydroxyacetone kinase,DAK)和自噬相关蛋白5-自噬相关蛋白12(Atg5-Atg12),进而干扰RIG-I/MDA-5信号级联,下调下游分子信号,包括干扰素β-启动子刺激因子1(interferon- β promoter stimulator 1,IPS-1)、诱导型I-κB优势激酶(IKKi)、肿瘤坏死因子受体相关因子3(TNF receptor-associated factor 3,TRAF-3)、TANK结合激酶1(TANK-binding kinase 1,TBK-1)等[60,62]。同时,DENV抗体复合物与FcR之间的结合可下调TLRs基因的表达,强烈刺激含TIR结构域的衔接子诱导干扰素-β(TRIF)和TNF受体相关因子6(TNF receptor-associated factor 6,TRAF-6)、SARM、TRAF家族成员相关NF-κB途径的激活因子(TANK)。TLR3、4和7与SARM和TANK呈负相关。SARM和TANK的上调导致TLR信号分子的下调[62,64],通过TLR信号途径介导的先天性反应受到抑制,促进病毒感染,从而将抗体介导适应性免疫与先天性免疫联系起来。
DENV有四种不同的血清型(DENV 1-4),不同血清型之间的交叉保护力较差,且感染一种血清型后再感染其他血清型的病毒,容易引发DHF/DSS,一种血清型的抗体容易引起异种血清型病毒的ADE[62,65]。另一方面,由于寨卡病毒(Zika virus,ZIKV)和DENV的同源性,两者血清存在交叉反应,导致两者中一种病毒的血清可促进另一种病毒的发展,存在ADE的风险[66]。
诱发病毒构象的变化从而促进感染也是ADE的重要机制。DENV的prM是膜(membrane,M)蛋白的前体蛋白,存在于未成熟DENV的表面。prM与E蛋白形成异二聚体,阻止病毒与宿主细胞膜的融合。在病毒成熟过程中,prM在低pH诱导下被弗林蛋白酶切割成M蛋白,使E蛋白发生重排,导致不具传染性的未成熟病毒颗粒成为成熟病毒并具有传染性。抗prM抗体可与prM结合,介导未成熟病毒与靶细胞的结合,从而增强病毒的传染性[67]。中东呼吸综合征冠状病毒(middle east respiratory syndrome coronavirus,MERS-CoV)发生ADE的机制之一是中和靶向冠状病毒S蛋白RBD的单克隆抗体介导的病毒进入。MERS-CoV的ADE与dpp4依赖的MERS-CoV遵循相同的进入途径。换句话说,RBD特异性中和单克隆抗体通过功能性模拟病毒受体介导冠状病毒进入,从而诱发ADE[68]。如果靶向中和抗体的其他部分不触发S蛋白的构象变化,则它们不太可能介导ADE。SARS-CoV-RBD单克隆抗体(S230)与SARS-CoV-RBD结合,引起SARS-CoV的构象改变[67]。可以推测,SARS-CoV可能具有与MERS-CoV相似的ADE机制。因此在开发疫苗时,特别是开发单克隆抗体药物时,需要尽量避免这些表位,防止诱发ADE。
Flipse等[69]研究发现,存在或不存在抗体的情况下,感染后结合和内化的DENV颗粒数量没有差异。然而,在ADE介导的DENV感染时,膜融合活性增强,增加疾病的严重程度。重要的是,在人原代巨噬细胞中未检测到抗体介导的免疫抑制信号。该研究为抗体介导的增强人原代巨噬细胞感染DENV的分子机制提供了新的视角。
6 疫苗开发与适应性免疫
疫苗的应用旨在诱导长寿命的抗原特异性记忆T细胞和B细胞,以期望这些T细胞和B细胞遇到同一病原体时介导快速、高亲和力的免疫反应。有些病毒容易变异,或存在免疫逃逸,这也是疫苗开发亟待解决的重要问题之一。
期望免疫产生的抗体提供针对病毒的保护,就需要找到对病毒有效的抗原表位。一旦鉴定并分离出此类抗体,就可以揭示病毒表面可被疫苗和免疫疗法靶向的区域,从而指导疫苗开发。如PRRSV、HIV和流感病毒等病毒容易变异,使得对其的防控十分困难,这就需要找到病毒有效且保守的细胞表位,从而为其提供有效的保护。HIV ENV具有许多重要的抗原决定簇,但这些抗原决定簇被埋在病毒突起的中央核心内,仅通过构象掩蔽暂时暴露,或CD4结合后才暴露[70-71]。有研究在HIV完整的ENV三聚体病毒刺突上展示多个表位,这些刺突经过改造来限制已知的非中和表位的暴露[72-73],或者将有效的抗原表位展示到蛋白质支架上,使抗体反应针对特定的位置。虽然这种改造过的三聚体确实能诱导更强的中和抗体滴度,但反应通常缺乏广谱性,诱导产生的抗体大多只针对自体免疫原。流感病毒最广泛有效的抗体已被定位在HA茎,表位聚焦的方法更加成功。可能的解释是,与HIV ENV相比,HA茎在整个病毒序列的保守程度更高,糖基化程度更低。将嵌合的HA茎部展示于免疫系统,免疫反应聚焦于保守的茎部,中和反应可能更有效,并包含一定程度的广谱性[74-75]。这对PRRSV和ASFV等疫苗的开发有借鉴意义。
T细胞的活化需要两种信号同时存在。第一信号,是由TCR与抗原肽-MHC复合物介导的抗原识别信号;第二信号,是由表达于T细胞表面的膜蛋白分子及其配体介导的共刺激信号。T细胞识别抗原后的完全活化须同时依赖两种信号通路。因此在调节T细胞中起关键作用的共刺激分子正成为免疫疗法中的重要分子。CD28可提供强大的协同刺激信号,增强CD28介导的共刺激可改善疫苗的免疫反应。当与活化的B细胞、DC或巨噬细胞上表达的配体B7分子结合时,CD28介导的共刺激信号会启动蛋白激酶B(protein kinase B,AKT)的活化,随后导致下游靶标核因子-κB(NF-κB)的活化,激活T细胞等的核因子[76]。此外,CD28还可诱导RAS的激活以及AKT、c-Jun N末端激酶和细胞外信号调节激酶(extracellular signal regulated dinase 1,ERK)的磷酸化,从而促进IL-2表达并促进T细胞的增殖和分化[77]。过表达CD28的转基因猪在免疫后CD4+T和CD8+T细胞显著增加,同时提高了针对PRRSV的中和抗体产生及IFN-γ的分泌[78]。这为解决PRRSV疫苗无法诱导高效T细胞反应提供新的思路。
NK细胞介导的抗原特异性记忆反应,使针对NK细胞的疫苗开发具有实际意义。改善NK细胞的ADCC,并使NK细胞产生抗原特异性记忆反应,成为流感病毒、HIV-1和其他病毒疫苗新的开发方向[79-81]。NK细胞的记忆性已发展到广泛的研究领域,利用NK细胞记忆反应的新型疗法和改进疫苗,让人充满期待。
7 总结与展望
适应性免疫和先天性免疫的边界越来越模糊,在机体抗病毒的过程中两者并不是独立存在,而是相互影响、协同作用。NK细胞在固有免疫和适应性免疫中都扮演重要角色,协调NK细胞与适应性免疫反应的关系,共同调节免疫功能,可成为开发新型免疫疗法和疫苗重要的思路。
在先天性免疫系统不能应对病毒感染时,机体就需要调用以T细胞和B细胞为主的适应性免疫系统清除病毒。为了对抗宿主的免疫反应,病毒进化出多种方式逃逸宿主的适应性免疫,如通过突变等方式影响中和抗体的产生与识别从而逃逸体液免疫,诱导Treg引发免疫抑制,上调PD-1/PD-L1的表达或抑制MHCⅠ/Ⅱ的抗原提呈作用等方式干扰、逃避T细胞的免疫应答。
同一病毒不同毒力的毒株会诱发不同的免疫反应。ASFV弱毒株体外感染DC或巨噬细胞时,能下调细胞表面MHCⅠ的表达,但强毒株无此效应。HP-PRRSV抑制猪外周血和肺中的Th17细胞功能的能力高于LP-PRRSV,可能与在感染早期CD4+T、CD8+T细胞显著降低有关。但耗尽CD8+T并没有影响PRRSV的感染。毒力差异如何影响病毒介导的细胞免疫反应,以及CD8+T细胞在PRRSV感染和清除中发挥什么作用仍需要进一步的研究解答。HP-PRRSV可诱导比经典PRRSV诱导更高水平的Treg。虽然已验证HP-PRRSV N蛋白可在体外诱导Treg,但鉴于N蛋白的高度保守性,可能N蛋白不是造成这种Treg诱导差异的原因。PRRSV的毒力基因nsp9、nsp10和高变性的nsp2或许是将来研究的方向。
流感病毒、ASFV和PRRSV等病毒的感染虽可以诱导抗体产生,但这类病毒的高变性使针对突变前抗原表位的中和抗体无法中和突变后的病毒。所以PRRSV疫苗难以对异源毒株攻击产生足够的保护。筛选出广谱性的细胞免疫与体液免疫抗原表位是下一步研究的重点。
人们对PRRSV、ASFV等病毒的免疫逃逸机制尚不完全清楚,阐明病毒编码蛋白的功能及其介导的免疫反应有助于我们理解病毒的致病和逃逸机制,指导防控产品的研发。
猜你喜欢
中国生殖健康(2020年2期)2021-01-18
温州医科大学学报(2019年4期)2019-04-28
中国医疗保险(2017年5期)2017-05-17
中国免疫学杂志(2017年1期)2017-01-17
中国康复理论与实践(2015年10期)2015-12-24
西南医科大学学报(2015年1期)2015-08-22
现代电生理学杂志(2015年1期)2015-07-18
畜牧兽医学报(2015年3期)2015-07-05
医学研究杂志(2015年6期)2015-07-01
癌变·畸变·突变(2015年3期)2015-02-27
