
急性孤立单侧眼内肌麻痹伴颅内病变的抗GQ1b抗体综合征1例并文献复习
2021-08-28 05:06陈丹阳李艳艳唐颖馨白霜王靖萱潘超唐洲平
神经损伤与功能重建 2021年8期
陈丹阳,李艳艳,唐颖馨,白霜,王靖萱,潘超,唐洲平
抗GQ1b抗体综合征(anti-GQ1b antibody syndrome)的概念由M Odaka等[1]在2001年首次提出,是由空肠弯曲菌、嗜血杆菌、巨细胞病毒等微生物通过分子模拟机制损伤中枢及外周神经系统,以在血清中可测得抗GQ1b抗体为重要特征的自身免疫性连续性疾病谱[1,2]。其主要临床表现为共济失调、腱反射消失、眼内、外肌麻痹、意识障碍等。该病可见于各个年龄段,患者症状复杂且常出现重叠现象,增加了临床分型的难度[3]。本文报道1例急性孤立单侧眼内肌麻痹伴颅内病变的抗GQ1b抗体综合征,伴血清抗GQ1b IgM抗体阳性,并进行相关文献复习,以提高对这类疾病的认识。
1 资料与方法
1.1 病例资料
患者,女性,40岁,因“急性发作右眼视物模糊1 d”入院。患者在1周前熬夜后出现上呼吸道感染症状,伴头晕和虚弱,无发热、头痛、腹泻等其他不适,休息后自行好转。昨日晨起时发现右眼视物模糊,左眼无异常,无视物重影,无眼睑下垂,无疼痛肿胀,无肢体活动障碍等其他不适。既往高血压病史,否认糖尿病及其他病史。入院后神经系统查体:神志清,高级皮质功能正常,双侧瞳孔不等大,右侧直径约4.0 mm,对光反射迟钝;左侧直径约1.5 mm,对光反射灵敏。右眼视力下降,视野无缺损。双侧眼球各向活动正常。余颅神经查体正常。四肢感觉、运动查体正常,腱反射等称引出,共济运动正常。
入院检查眼底结果正常。血常规、粪常规、凝血4项、肿瘤标志物、甲状腺功能全套均未见明显异常。尿液常规分析结果显示:红细胞1+,尿葡萄糖+。血生化全套结果显示:总胆固醇6.47 mmol/L,甘油三酯2.40 mmol/L,低密度脂蛋白4.64 mmol/L,尿酸370.7 umol/L,同型半胱氨酸15.1μmol/L,葡萄糖7.24 mmol/L,糖化血红蛋白6.1%。自身抗体筛查提示抗核抗体阳性(核均质型,1∶100)。急查头部CT未见明显异常;头颅磁共振成像(magnetic resonance imaging,MRI)检查示:左侧海马及丘脑区见小片状长T1长T2高T2液体衰减反转恢复序列(fluid-attenuated inversion recovery,FLAIR)信号灶,弥散加权成像(diffusion weighted imaging,DWI)上呈高信号,增强未见明显强化,见图1;头颈部CT血管成像(computed tomography angiography,CTA)检查未见明显异常。患者头部MRI表现与脑梗死病灶相似,但临床症状与病灶不符,考虑肿瘤、炎性脱髓鞘性、免疫性等疾病可能。进一步完善检查,视觉诱发电位(visual evoked potential,VEP)未见明显异常;体感诱发电位(somatosensory evoked potentials,SEP)结果显示双下肢感觉通路传导障碍。脑脊液检查和颈髓、胸髓MRI未见明显异常。入院第6天同时送检患者的血与脑脊液标本,结果显示:血清抗人神经节苷脂抗体检测显示抗GQ1b IgM抗体滴度升高(++),IgG抗体滴度正常,而脑脊液抗GQ1b IgG及IgM抗体滴度均正常,见图2;其他血与脑脊液神经节苷脂抗体(GT1b、GD1a、GD1b、GM1、GM2、GM3)和脱髓鞘抗体(NMO、MBP、MOG)均为阴性。
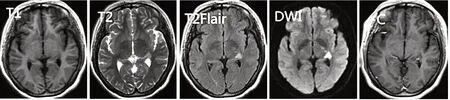
图1 本例患者头部MRI影像

图2 抗GQ1b抗体阳性神经节苷脂抗体谱图
本例最终诊断为抗GQ1b抗体综合征,临床分型为不完全形式的Miller-Fisher综合征(Miller-Fisher syndrome,MFS)(急性瞳孔散大亚型)。诊断依据:①抗GQ1b抗体综合征的确诊依据为血清抗GQ1b抗体阳性[4]。②符合急性瞳孔散大亚型的诊断标准:根据新的分类标准[5],MFS各亚型的确诊需要符合相应的临床特征关键点(急性瞳孔散大亚型的临床特征关键点为麻痹性瞳孔散大、无共济失调及无嗜睡)并存在支持依据(血清抗GQ1b抗体阳性),且需除外其他疾病。本例无意识障碍、锥体束受损体征,头颅MRI未提示脑干异常信号,故排除Bickerstaff’S脑干脑炎(Bickerstaff’s brainstem encephalitis,BBE)及脑干梗死的诊断;无肉毒杆菌流行病学史,且不伴相关中毒症状,故排除肉毒杆菌中毒的诊断;无Wernicke脑病眼球运动障碍、小脑性共济失调、精神意识障碍的典型临床三联症及影像学表现,故排除Wernicke脑病的诊断。
给予患者静脉注射丙种球蛋白5 d(400 mg/kg/d),其右眼视物模糊症状好转,但右眼瞳孔直径仍大于左侧。患者出院后口服醋酸泼尼松片继续治疗。3个月后患者再次入院复查,视物模糊症状消失,偶有头昏,无其他特殊不适,查体双侧瞳孔不等大,右侧3.0 mm,左侧1.5 mm,余神经系统查体未见异常。复查神经节苷脂抗体显示,血清抗GQ1b IgM抗体阳性(+),滴度较3月前减低。继续随诊。
1.2 方法
搜集文献并分析。使用Pubmed数据库,输入关键词“anti-GQ1b antibody和isolated mydriasis或isolated internal ophthalmoplegia”,检索时间截止至2020年12月8日,对检索结果逐篇人工核查,获得所有表现为孤立眼内肌麻痹(不伴眼外肌麻痹)的抗GQ1b抗体综合征的病例报道。对符合条件的病例,从性别、发病年龄、临床分型、前驱感染类型、症状、实验室检查、影像学检查、治疗及预后等各方面进行描述性研究分析。
2 结果
共获得7篇符合条件的文献[6-12],包含7例患者,平均年龄33岁(21~53岁),男女比例为4:3。①临床分型:4例为MSF,1例为BBE,2例未明确分型。②前驱感染类型:6例患者存在前驱感染,其中3例为呼吸道感染,2例为腹泻,1例存在不明原因的前驱发热症状。③症状:6例主要表现为孤立的双侧眼内肌麻痹而无眼外肌麻痹;1例以孤立的眼内肌麻痹为独立首发症状,随着病程进展出现眼外肌麻痹及MFS其他典型症状;3例出现共济失调;5例腱反射减弱,1例腱反射增强,1例腱反射正常;6例对光反射减弱,1例对光反射情况不详;其他伴随症状包括头晕、头痛等。④实验室检查:7例患者均进行脑脊液检查,其中1例出现脑脊液蛋白-细胞分离现象;7例均有血清抗GQ1b IgG抗体阳性。⑤影像学检查:5例接受头颅MRI检查,结果均未见明显异常。⑥治疗及预后:1例接受免疫吸附血浆净化治疗,1例接受静脉注射丙种球蛋白5 d[400mg/kg/d]治疗,2例接受激素冲击治疗,3例未接受治疗。7例患者均完全康复,预后较好。
3 讨论
抗GQ1b抗体综合征的说法最早由M Odaka等[1]在2001年首次提出,包括MFS、有眼外肌麻痹的吉兰-巴雷综合征(Guillain-Barre syndrome,GBS)、BBE和急性眼肌麻痹。抗GQ1b抗体综合征的头部MRI检查多正常,仅在少数患者的T2加权像中可出现一些高信号的异常影像。绝大多数抗GQ1b抗体综合征患者存在前驱感染症状,如上呼吸道感染症状,腹泻[13],其中以上呼吸道感染多见[1,14]。神经节苷脂是一类含有唾液酸的鞘糖脂,参与细胞膜形成,具有稳定胞膜、调节突触传递、调节神经细胞增殖与分化等重要的生物学功能[15,16]。神经节苷脂根据含己糖的数目及位置不同,分为GM1、GD1a、GT1a、GQ1b、GT1b、GM2、GM3等,在体内分布广泛,在神经系统含量最为丰富。不同类型的神经节苷脂在神经系统的分布有特定区域性,其中GQ1b主要分布在动眼神经(Ⅲ)、滑车神经(Ⅳ)、展神经(Ⅵ)的节旁髓鞘,神经肌肉接头,四肢肌梭和脑干网状结构[17]。现研究认为,抗GQ1b抗体综合征的主要发病机制为分子模拟[2],由于前驱感染时入侵机体的微生物存在的抗原与宿主的某些抗原具有相似结构,导致机体发生免疫交叉反应,产生针对体内特定神经节苷脂位点的抗体,从而损伤神经系统。综合疾病发生的机制及GQ1b在神经系统的分布特点,我们可以推断,抗GQ1b抗体综合征的临床表现主要为眼肌麻痹(Ⅲ、Ⅳ、Ⅵ对脑神经)、共济失调、腱反射消失(肌梭损伤)、四肢乏力(神经肌肉接头损伤)、意识障碍(脑干网状结构)等。
随着对这类疾病的认识增加,Shahrizaila N等在2013年时提出将抗GQ1b抗体综合征分为以下3个亚型:Fishere-Bickerstaff综合征,咽-颈-臂无力以及交叉重叠亚型。其中Fishere-Bickerstaff综合征又可分为MFS、BBE这2种亚型。其中MFS是一类以急性的眼肌麻痹,共济失调及腱反射消失的三联征为主要临床特征的疾病,由Miller Fisher在1956首次提出,其年发病率估计值不到百万分之一[1,18]。Wakerley等[5]于2014年提出MFS新的分类标准,将MFS分为2种形式:典型MFS(急性眼外肌麻痹、共济失调和腱反射消失)和不完全形式的MFS(急性眼外肌麻痹、急性上共济失调性神经病、急性瞳孔散大和急性眼睑下垂)。该病的平均发病年龄为43.6岁[19]。由于部分MFS患者可发展为GBS,故MFS常被认为是GBS的变异型[20]。约90%以上的MFS患者可在急性期检测到血清中的抗GQ1b抗体阳性,且常表现为抗GQ1b IgG抗体阳性[21]。发生MFS时,眼肌瘫痪包括眼外肌瘫痪(眼球运动异常、复视)和眼内肌瘫痪(瞳孔改变),两者可同时出现,也可单独出现[22,23]。既往的病例有单独眼外肌瘫痪不伴瞳孔异常的报道,亦有单独瞳孔散大不伴眼外肌受累的报道[24]。值得注意的是,MFS多表现为双侧对称性眼肌瘫痪,单侧眼肌瘫痪少见[25-27]。由此可见,本文报道的抗GQ1b抗体综合征患者,以急性孤立单侧眼内肌麻痹为主要临床表现,伴有颅内病变,且血清抗GQ1b IgM抗体阳性,比较罕见,有很大的学习价值。
本例临床表现为单侧的视物模糊及孤立的眼内肌麻痹。由于MRI结果显示左侧丘脑区存在病灶,故推测,视物模糊可能是由于外侧膝状体损伤,右侧视觉传导通路受损所致,而患者视觉诱发电位正常,可能是由于VEP本身对视交叉后的病变不敏感。而眼内肌麻痹的症状,是由于动眼神经睫状神经节受损所致[10]。在之前的1项包含581例表现为急性眼肌麻痹和共济失调患者的研究中,466例被诊断为了MFS,其中1%的MFS患者出现了脑MRI异常[28]。在另一项研究中,5%~23%的MFS患者出现躯体感觉诱发电位(SEPs)异常[29]。电生理检查敏感度较高,可以发现早期亚临床症状,本例患者SEPs显示双下肢感觉通路传导障碍,考虑是由于肌梭受累,导致深感觉传导阻滞,但该病例尚未出现相关临床症状。此外,IgM常被视为近期感染的标志,IgG被视为既往感染的标志。但在本例中,患者入院第6天检测显示血清抗GQ1b IgM抗体滴度升高(++)。出院3个月后复查,虽然滴度较首次检查时降低,但血清抗GQ1b IgM抗体仍为阳性(+),且2次检查抗GQ1b IgG抗体均为阴性。这一现象可能与前驱感染类型的不同有关。B Schwerer等[30]发现,MFS患者体内抗GQ1b抗体的类型(IgG或者IgM)可能取决于前驱传染源的类型(呼吸道感染或者消化道感染)。但是,此项研究中患者数量相对较少,数据有限,这一结论仍需更大规模的临床数据去验证。本例可提高之处在于应补充新斯的明试验等检查,排除重症肌无力等其他疾病,使诊断更加严谨。
抗GQ1b抗体综合征具有自限性,有良好的预后,症状较轻的患者可通过保守治疗而得到完全改善。由于抗GQ1b抗体综合征和GBS有相似的发病机制,因此临床上对伴有严重并发症的抗GQ1b抗体综合征常采用GBS的治疗方法。JR Overell等[11]认为,对伴有严重共济失调、严重延髓麻痹、呼吸无力、肢体无力等并发症的MFS患者,应及时采取治疗GBS相同的方法,即及时进行丙球及血浆置换治疗。但目前这一结论仍缺乏随机临床对照研究的证据。本例患者病情复杂,我们对其进行静脉注射丙种球蛋白治疗,并取得了良好的疗效。
综上所述,抗GQ1b抗体综合征具有临床表现复杂,分型困难的特点。本例急性孤立单侧眼内肌麻痹伴颅内病变及抗GQ1b IgM抗体阳性的病例为首次报道。这一发现有助于加深临床工作者对该疾病的认识,减少误诊、漏诊的发生率。