
原发性低钾型周期性麻痹分子遗传学及其治疗的研究进展
2021-08-24 02:51:30王衡钟敏
山东医药 2021年25期
王衡,钟敏
1重庆医科大学附属儿童医院神经内科 国家儿童健康与疾病临床医学研究中心儿童发育疾病研究教育部重点实验室,重庆400014;2儿科学重庆市重点实验室
周期性麻痹是一组以周期性弛缓性肌无力为主要表现的骨骼肌离子通道疾病,包括低钾型周期性麻痹(HypoPP)、正常钾型周期性麻痹(NormPP)、高钾型周期性麻痹、Andersen-Tawil综合征。其中,HypoPP在周期性麻痹中最常见,发病率约为1/10万[1],分为原发性和继发性。原发性HypoPP是一种罕见的常染色体显性遗传病,由编码骨骼肌钠、钙、钾离子通道基因突变引起的,通常于10~20岁发病,临床表现为局灶性或全身性弛缓性肌无力和低钾血症(<3.5 mmol/L)[2]。大多数原发性HypoPP是由骨骼肌L型Cav1.1通道基因(CACNA1S)或骨骼肌Nav1.4通道基因(SCN4A)α亚基突变引起的。我国曾报道由编码电压门控钾离子通道的KCNJ18基因突变引起的散发性HypoPP,东莞地区还发现G116A、G119A、C167A、A745G、G576C五个新的错义突变位点[3]。其中,约60%原发性HypoPP与CACNA1S突变有关,定义为HypoPP-1型(OMIM:114208);约20%原发性HypoPP与SCN4A突变有关,定义为HypoPP-2型;其余原发性HypoPP不存在这两个基因突变,归为HypoPP-3型[4-5]。目前,原发性HypoPP的分子遗传学机制仍不完全清楚,其诊断共识和治疗指南尚无统一意见,治疗措施主要是避免诱因和补充钾剂。本文结合文献就原发性HypoPP分子遗传学及其治疗的研究进展作一综述。
1 原发性HypoPP的分子遗传学机制
原发性HypoPP以发作性肌无力伴血钾降低、补钾后肌无力迅速缓解为特征。目前认为,HypoPP是由CACNA1S或SCN4A突变所致的氨基酸改变,致使骨骼肌L型Cav1.1通道或Nav1.4通道中电压传感器跨膜段的一个门控电荷改变而形成门控孔电流(也称ω电流),阳离子在静息状态下通过该孔渗漏,产生异常静息电位,导致肌细胞膜对神经刺激反应减弱,收缩力降低,从而引起肌无力[6]。
1.1 CACNA1S突变 CACNA1S定位于人染色体1q31-32上,全长约73 kb,包含44个外显子,编码骨骼肌细胞二氢吡啶受体敏感的L型Cav1.1通道蛋白。该通道蛋白是由5个亚基组成的寡聚蛋白,即两个高分子量多肽亚基(α1、α2)和三个较小的亚基(β、γ、δ),其中α1亚基是主要的功能单位。HypoPP-1型是由编码二氢吡啶受体α1亚基(Cav1.1α1)错义突变引起的。二氢吡啶受体是一种锚定在骨骼肌纤维管状膜上的蛋白复合物,由4个同源结构域(Ⅰ~Ⅳ)组成,每个结构域包含6个跨膜区段(S1~S6)。其中,S4区段中的精氨酸通过与相邻节段的残基相互作用,形成了一个非常狭窄的隔膜,能够将无离子通透性的细胞内和细胞外的水隙分开。大部分基因突变影响结构域Ⅱ~Ⅳ的S4区段,该区段中的精氨酸被中性残基替代,破坏了这种结构,桥接了细胞内外液体,形成了一个可传导Ca2+并响应去极化而打开的门控通道,从而控制肌浆网Ca2+释放,影响肌肉的兴奋—收缩偶联[7]。WU等[8]研究发现,Cav1.1通道蛋白STAC3过表达能够增强非洲爪蟾卵母细胞膜上Cav1.1表达,在非肌肉来源的细胞中实现对HypoPP突变通道的功能评估。目前认为,周期性麻痹(HypoPP、NormPP)、恶性高热综合征、与Cav1.1相关的肌病和肌强直性营养不良1型四种肌肉疾病,可能与CACNA1S突变或剪接缺陷有关,以HypoPP最常见[9]。
与原发性HypoPP相关的CACNA1S突变导致的氨基酸位点改变主要包括R528H/G/C/L、R1239H/G、R897S/M、R900G/S、H916Q、V876E、R489H、R1242G,其 中R528H和R1239H最 常 见[5,10-18]。MILLER等[10]研究报道,c.1583 G>A(p.Arg528His)突变所致的原发性HypoPP患者首发年龄为(14±5)岁,而c.3716 G>A(p.Arg1239His)突变为(7±4)岁,表明c.3716 G>A(p.Arg1239His)突变所致的原发性HypoPP发病年龄更早。HIRANO等[11]报道了一例CACNA1S基因c.2698 A>G(p.Arg900Gly)突变的男性周期性麻痹发作患者,其基因突变发生在结构域Ⅲ的S4区段,患者在肌无力恢复后血钾明显升高,这可能是因为发作间期低钾血症使肌膜不稳定,从而向血清释放肌内钾。CHABRIER等[12]报道了一例氨基酸R897S变化的先证者,患者是土耳其血统近亲结婚夫妇的独生子女,无肌肉疾病家族史,1岁左右发病,表现为以下肢为主的全身性肌无力,发作持续时间10~30 min,该突变表明结构域Ⅲ的S4区段功能障碍也可引起原发性HypoPP,故分子研究不应局限于结构域Ⅱ、Ⅳ的S4区段。WEBER等[13]报道了一例由CACNA1S突变导致的女性原发性HypoPP患儿,首发年龄为3岁,在中等强度运动或感染诱发下表现出明显的下肢无力甚至不能行走,经鉴定是CACNA1S c.3715 C>G(p.Arg1239Gly)突变引起的。c.2627 T>A(p.Val876Glu)突变最早在一个南美家系中被发现,该家系共发现6例原发性HypoPP患者[14],之后分别于2015年在一例中国女性[15]和2020年在一个日本家系[16]中发现该位点突变。目前,c.2690 G>A(p.Arg897Met)突变只在中国人群中报道,是在一例16岁男性原发性HypoPP患者中首次发现,CACNA1S的突变位点为R897M,患者母亲亦存在该位点突变且已发病,经补钾治疗有效[17]。总之,CACNA1S突变位点可发生在多个结构域的不同区段,并且某些突变位点,如R528H、R1239H,可表现为不完全外显率,但更多的突变位点有待于进一步探索。CACNA1S常见的突变位置以及核苷酸、氨基酸变化见表1。
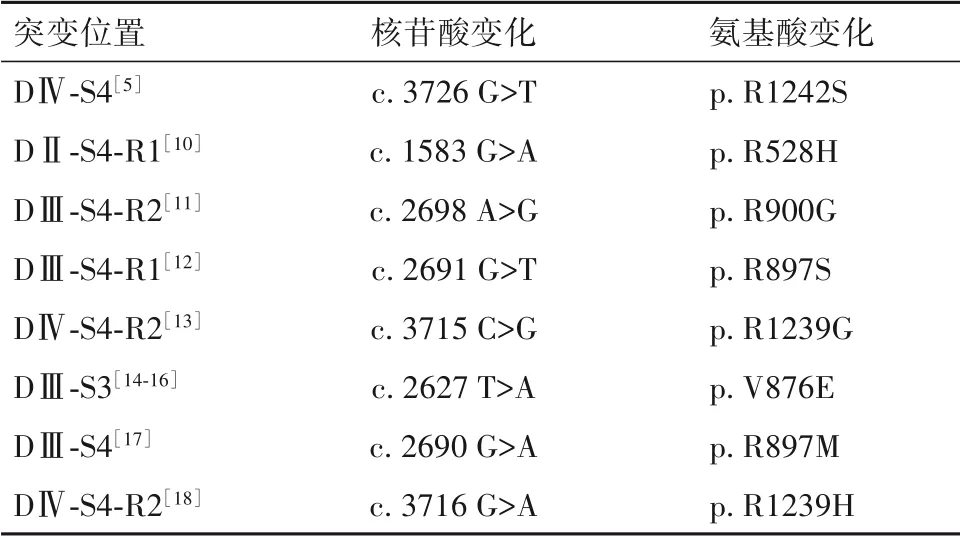
表1 CACNA1S常见的突变位置以及核苷酸、氨基酸变化
1.2 SCN4A突变 SCN4A定位于人染色体17q23-25,全长35 kb,有24个外显子,编码骨骼肌电压门控钠通道Nav1.4α亚基。Nav1.4α亚基由围绕中心离子孔的4个同源结构域(Ⅰ~Ⅳ)组成,每个结构域由6个跨膜片段组成。SCN4A突变可导致高钾型周期性麻痹、HypoPP-2型(OMIM:613345)、先天性副肌强直和先天性肌无力综合征。
JIANG等[19]构建了细菌钠通道模型,阐明通过门孔的离子渗透是由电压传感器状态和R4的转子构象动态变化控制的,这在分子水平上揭示了原发性HypoPP的发病机制。电压门控钠通道Nav1.4电压传感器的S4跨膜区段中带正电荷的残基单个突变可引起原发性HypoPP。最外层门控电荷(R1、R2)突变会在电压传感器中形成病原性门控孔,阳离子在静息状态下通过该孔泄漏,产生门控孔电流,从而导致原发性HypoPP。钠通道是一种高选择性电压门控通道,通过构象变化激活去极化,然后失活阻断钠电流,引起膜复极化。该通道失活的轻微延迟即可导致肌强直或肌无力,在运动后休息、摄入碳水化合物或胰岛素、感染等诱发因素下,电压门控钠通道长时间处于膜去极化(从-90 mV至-60 mV)而失活状态[2,9]。
有研究报道,原发性HypoPP-2型的致病性突变多位于SCN4A编码的精氨酸碱基上,特别是在结构域Ⅱ上。SCN4A突变的主要位点包括R222W/Q、R225W/G、R669H/G、R672H/G/S/C、R675Q、R1129Q、R1132Q/G、R1135H/C、P1158S和N820Y,其中R672G和R1132Q最常见[5,20-28]。CARLE等[20]于2006年在一个欧洲家系中发现了c.3395 G>A(p.Arg1132Gln)突变,该突变导致钠离子通道Nav1.4α亚基结构域Ⅲ电压传感器S4区段中的精氨酸被谷氨酰胺取代,改变了钠通道激活的电压依赖性,增强了快速和慢速失活。c.2458 A>T(p.Asn820Tyr)突变是2020年在一个中国家系中报道的新发突变,因SCN4A第14外显子突变,位于Nav1.4α亚基结构域Ⅱ的S6区段末端,该突变可导致Nav1.4静息电位持续去极化,从而降低了肌纤维的兴奋性,主要表现为发作性四肢无力[21]。c.3386 G>A(p.Arg1129Gln)突变是2010年在一个中国家系中发现的,是SCN4A结构域Ⅲ电压传感器S4区段的精氨酸被谷氨酰胺取代,该家系中共发现3例HypoPP、5例NormPP,氯化钾能够缓解HypoPP患者肌无力症状,但对NormPP患者无效,提示钠通道功能障碍可能与R1132Q突变有关[22]。c.2024 C>T(p.Arg675Gln)突变在两个中国家系中被发现,位于SCN4A编码的骨骼肌电压门控钠离子通道Nav1.4α亚基结构域Ⅱ的S4区段,该区段第3个带正电荷的精氨酸被疏水氨基酸残基取代,在电压传感器中形成病原性门控孔,从而导致异常门控孔电流形成,患者伴有肌肉酸痛和肌酸激酶升高,肌肉酸痛和肌酸激酶升高可能与该位点基因突变密切相关[5]。c.664 C>T(p.Arg222Trp)是与原发性HypoPP相关的第一个引起骨骼肌钠通道Nav1.4结构域Ⅰ改变的突变,在既往报道该突变的两个家系中可出现呼吸功能不全等严重临床表现[23],而在BAY⁃LESS-EDWARDS等[24]报道的家系中则具有以四肢无力为特征的轻度临床表现,通过分子动力学模拟证实,R222W突变产生的钠通道失活缺陷是骨骼肌纤维动作电位衰减的主要驱动力,而由该位点突变产生的超极化诱导ω电流则能促进肌纤维去极化。KOKUNAI等[25]研究报道,SCN4A第4外显子杂合状态下新的错义突变位点c.611 C>A(p.Ala204Glu),该突变导致骨骼肌电压门控钠离子通道Nav1.4α亚基结构域Ⅰ的S3区段改变,乙酰唑胺与补钾能够减少患者肌无力发作。R669G是荷兰报道的SCN4A新突变[26],该研究从234个不相关的家系中鉴定了405例骨骼肌通道病患者,结果发现该突变导致荷兰非营养不良性肌强直和周期性麻痹的患病率分别为1.7/10万、0.69/10万。SCN4A常见的突变位置以及核苷酸、氨基酸变化见表2。

表2 SCN4A常见的突变位置以及核苷酸、氨基酸变化
2 原发性HypoPP治疗
原发性HypoPP的临床诊断并不困难,结合基因检测可进一步明确病因,但目前缺乏有效的根治方法。
原发性HypoPP导致的肌无力是可逆的,持续治疗可减少肌无力发作频率,并防止永久性肌无力发生。有研究报道,一例女性原发性HypoPP患者经螺内酯、补钾等治疗,从轮椅依赖者转变为在没有帮助下步行甚至慢跑,表明中重度肌无力发作经适当治疗是可以逆转的[13,29]。原发性HypoPP的治疗管理主要基于三个原则:第一,改变生活方式,尽量减少HypoPP的触发因素;第二,急性期稳定血钾水平;第三,使用碳酸酐酶抑制剂等预防肌无力发生。
2.1 减少触发因素 原发性HypoPP常见的触发因素及其潜在机制:①高碳水化合物饮食、葡萄糖输液、糖皮质激素等,这些因素能够使胰岛素介导的Na+-K+-ATP酶活性增强,通过阻碍钾离子通道活性,抑制钾离子外流;②剧烈运动后休息,长时间或过度运动可能导致局部酸中毒,夜间或清晨休息时肌细胞内钾流入增多;③创伤和情绪压力,能够使儿茶酚胺介导的Na+-K+-ATP酶活性增强,通过阻碍钾离子通道活性,抑制钾离子外流。
2.2 急性期稳定血钾水平 目前,原发性HypoPP常见的治疗药物主要有钾补充剂、碳酸酐酶抑制剂、利尿剂等。其中,碳酸酐酶抑制剂乙酰唑胺、二氯苯酰胺临床治疗HypoPP已有近50年历史,能够有效降低HypoPP发作频率,提高患者生活质量[30]。但碳酸酐酶抑制剂不良反应较多,如感觉异常、疲劳、肌肉痉挛和轻度可逆性认知功能障碍等[14]。利尿剂布美他尼是目前尚处于研究阶段的治疗药物,可通过抑制Na+-K+-2Cl-协同转运体,限制与重复动作电位相关的细胞内氯离子浓度升高,从而导致肌纤维膜在低细胞外钾状态下稳定。虽然布美他尼是HypoPP的潜在治疗药物,但尚未在临床上应用,还需要进一步研究[31-33]。
急性发作期治疗主要采取口服或静脉补钾。口服补钾速度一般为0.4~0.8 mEq/(kg·h),每天不超过200~250 mEq。当无法口服补钾时需要静脉补钾,补钾速度一般不超过20 mEq/h,每天不超过200 mEq。
2.3 预防肌无力发生 预防肌无力发生的药物治疗:①乙酰唑胺:成人125~1 000 mg/d,儿童5~10mg/(kg·d),能够预防HypoPP肌无力发作,但使用乙酰唑胺可增加肾结石的发生风险[30],故在使用乙酰唑胺预防治疗前应先进行肾脏超声检查;②补钾:口服补钾每天30~60 mEq,首选缓释制剂;③保钾利尿剂:如氨苯蝶啶50~150 mg/d、安体舒通25~100 mg/d、依普利酮50~100 mg/d,这些保钾利尿剂不仅能预防HypoPP肌无力发作,还能使肌纤维复极化增强和肌力增加;④二氯苯酰胺:已被批准用于预防HypoPP肌无力发作,能够降低肌无力严重程度、发作频率,减少发作持续时间,但其具体治疗剂量尚不统一。
综上所述,原发性HypoPP的分子遗传学机制主要是由于CACNA1S或SCN4A突变致使电压门控钙通道Cav1.1或钠通道Nav1.4改变而产生ω电流,继而影响膜结构域Ⅱ~Ⅳ的S4区段,最终出现局灶性或全身性弛缓性肌无力和低钾血症。原发性HypoPP的临床诊断并不困难,结合基因检测可进一步明确病因,但目前缺乏有效的根治方法,治疗原则主要为减少触发因素、急性期稳定血钾水平以及预防肌无力发生。原发性HypoPP的常规治疗药物主要有钾补充剂、碳酸酐酶抑制剂、利尿剂等。但由于国内外原发性HypoPP的病例报道不多,许多药物的有效性和安全性尚缺乏证据。未来需要进一步探索原发性HypoPP的分子遗传学机制,以便研发安全有效的治疗药物。
猜你喜欢
中外医疗(2022年31期)2022-04-03 06:42:44
世界最新医学信息文摘(2020年68期)2020-12-25 11:55:27
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:30
国际放射医学核医学杂志(2020年2期)2020-05-30 12:39:56
中国中医急症(2019年10期)2019-05-21 07:20:42
中国社区医师(2017年34期)2018-01-06 00:53:12
现代检验医学杂志(2016年4期)2016-11-15 02:00:58
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:13
护理实践与研究(2015年1期)2015-08-05 03:36:50
中国实用乡村医生杂志(2014年9期)2014-01-27 04:51:27
