
神经元核内包涵体病1例的影像诊断报道并文献复习
2021-08-24 08:20金晓梅刘晓蓉王贤慧翟万庆通讯作者
影像研究与医学应用 2021年14期
金晓梅,刘晓蓉,王贤慧,周 亦,翟万庆(通讯作者)
(太仓市第一人民医院<苏州大学附属太仓医院>神经内科 江苏 太仓 215400)
神经元核内包涵体病(NIID)是一种以中枢和周围神经系统神经元细胞核内嗜酸性透明包涵体形成为特征的慢性进展性神经变性疾病。皮肤、胃肠道及腓肠神经活体组织检查可见同样的包涵体。包括散发型及家族型,儿童、少年及成人均可患病。临床症状具有高度异质性,磁共振(MRI)弥散加权成像(DWI)上皮髓交界区高信号是本病的特征性影像表现。本文报道了我院1例记忆力下降为主要临床特征的NIID患者,结合其典型影像学变化,并复习相关文献,旨在加强临床及影像医生对NIID的认识,降低误诊漏诊率。
1 病例介绍
患者男性,55岁,因“记忆力下降5天”于2018年12月25日入院。患者入院前5天无明显诱因出现记忆力下降,短时记忆及长时记忆均受影响,时间、空间记忆及定向较差。主要表现为无法回忆之前及刚刚发生的事,无法识别方向,找不到熟悉的目的地。无言语及行为异常,无肢体活动障碍,无头晕复视,无头痛呕吐,无进食进水呛咳,无畏寒发热,无意识障碍,无肢体抽搐,无尿便障碍,无皮肤出汗异常。发病以来饮食睡眠可,二便通畅,近期体重无明显变化。于我院门诊查头颅CT(西门子16排螺旋CT)提示双侧额叶缺血灶(图1),颈超(Hitachi Aloka Arietta 70型超声诊断仪)提示右侧颈总动脉分叉处斑块形成,拟“脑梗死”收住入院。
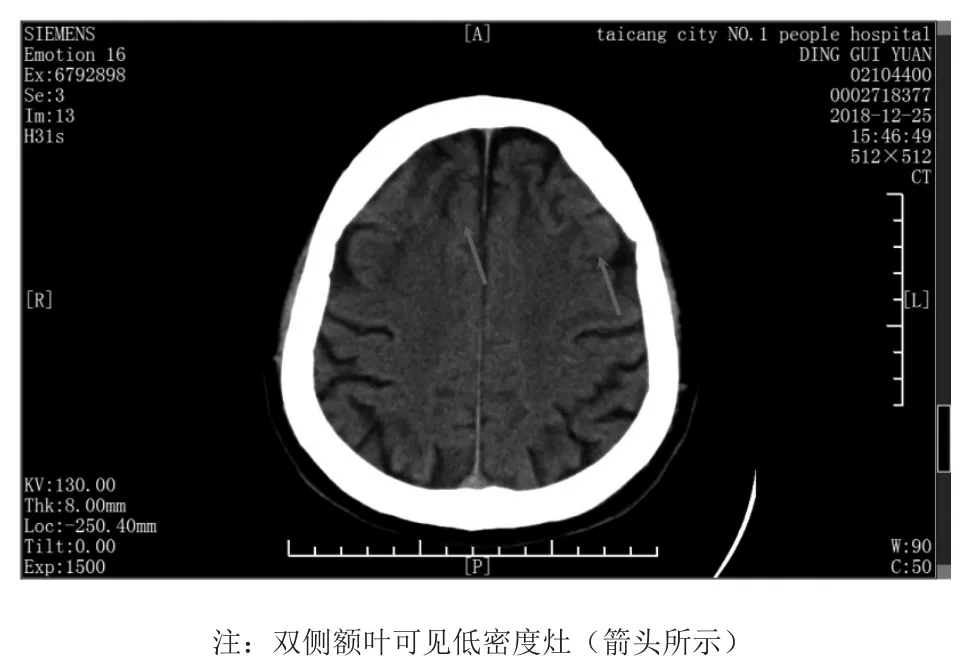
图1 患者CT结果
既往史:既往否认高血压、糖尿病、冠心病、脑卒中等慢性病史,否认肝炎、结核、梅毒等传染病史及特殊感染史,否认慢性毒物接触史,无特殊用药史,家族中无类似病史,少量吸烟饮酒史。
入院查体:生命体征及内科查体无殊。神经系统查体:神志清楚,精神可,口齿清,可按指令动作,定时、定向、记忆力下降,计算力下降,MMSE评分14分。双侧瞳孔等大等圆,直径3.0 mm,对光反应灵,双侧额纹、鼻唇沟对称,双耳概测听力可,咽反射存在,伸舌居中,颈软,无抵抗感,四肢均可活动,肌力5级,肌张力不高,双侧肢体及面部痛温觉对称存在,指鼻实验、跟膝胫实验正常,Romberg征(-),双侧巴氏征阴性,膝反射(++),跟腱反射(++),肌阵挛(-),布氏征、克氏征阴性。
辅助检查:血常规、粪常规、尿常规、甲功三项、凝血功能、男性肿瘤指标、叶酸、维生素B12、同型半胱氨酸、肌钙蛋白、肌红蛋白、B型脑钠肽、输血前常规正常。生化:甘油三酯2.96 mmol/L,谷草转氨酶45.5 U/L,葡萄糖6.44 mmol/L,稍高。自身抗体初筛:抗核抗体HEP2(核仁型),阳性(1:100),余阴性。动态心电图、心脏超声正常。头颅磁共振扫描(GE-Discovery GE-3.0T)(2018-12-27):双侧额颞叶、枕叶皮髓质交界处磁共振弥散加权成像(DWI)异常高信号(图2),复查头颅磁共振扫描(2019-01-02)DWI病灶较前相仿,增强扫描(2019-01-02)无明显异常。
诊治经过:患者主要表现为急性起病的皮层症状,结合其烟酒嗜好及门诊颈超提示动脉粥样硬化,入院后定位于皮层,定性优先考虑急性脑血管病,门诊头颅CT排除出血,考虑缺血性脑血管病,症状持续性,考虑脑梗死,TOAST分型考虑大动脉粥样硬化型。入院后予以卒中二级预防(阿司匹林、阿托伐他汀),改善侧支循环(丁苯酞),改善认知(多奈哌齐),活血化瘀,清除自由基等对症治疗,并完善相关检查了解有无其他致病原因。患者头磁共振检查DWI相提示双侧额颞叶、枕叶皮髓质交界处DWI异常高信号(见图2),结合病灶特点,不支持急性脑梗死诊断,且患者无特殊感染及中毒史,入院后一系列辅助检查不支持炎症性、感染性、中毒、肿瘤等疾病,患者影像学符合神经元核内包涵体病的典型影像学表现,建议患者至上海华山医院进一步诊治,于华山医院完善小腿皮肤活检(右侧下肢外侧,包括表皮、真皮和一定的皮下组织),标本送至北京天坛医院,病理结果显示:部分脂肪细胞、汗腺细胞、纤维细胞的细胞核内见P62抗体及泛素抗体染色阳性的包涵体,结合临床诊断,神经元核内包涵体病(图3),给予辅酶Q10治疗,现随访患者记忆力较前好转,症状未反复。
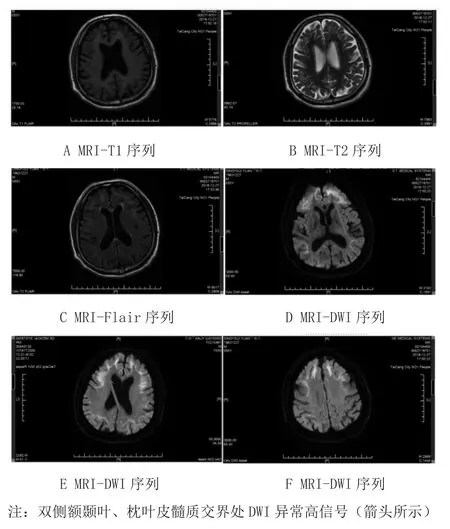
图2 患者入院MR结果
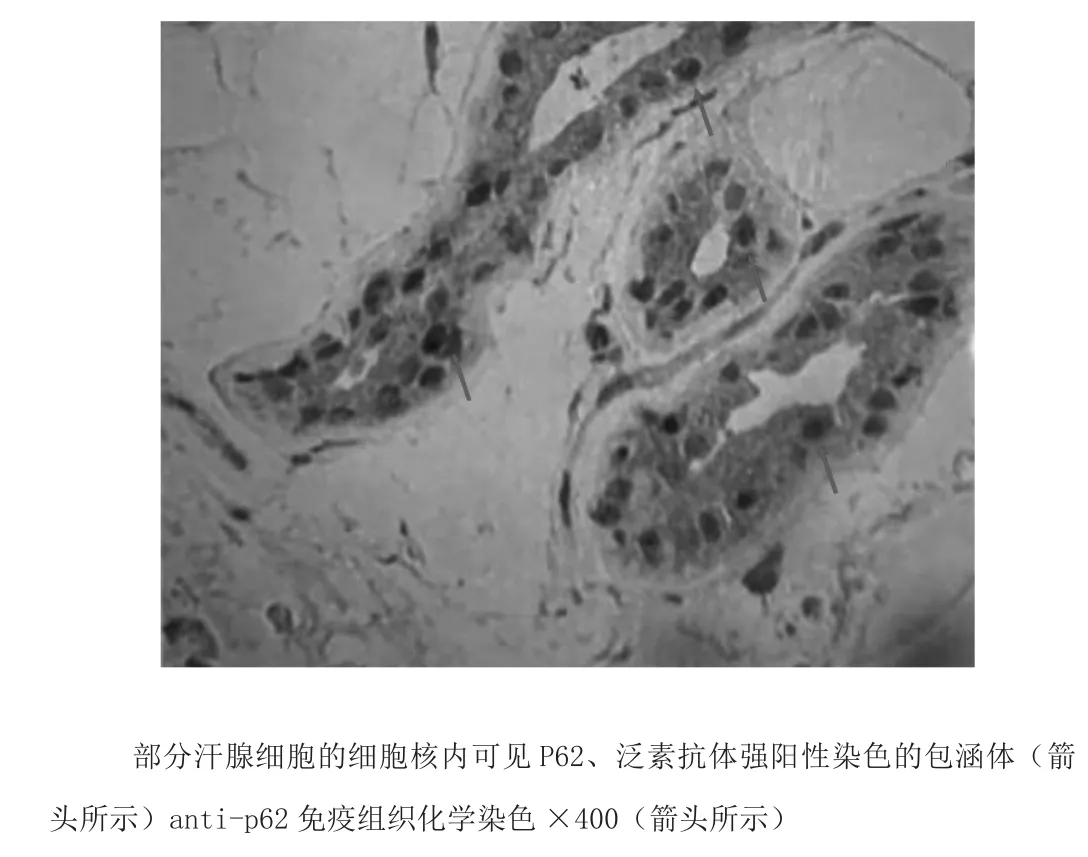
图3 患者皮肤病理学改变
最终诊断为散发型成人型神经元核内包涵体病。
2 讨论
本例患者主要表现为认知功能障碍,无周围神经系统损害表现及自主神经功能障碍,入院后一系列临床和实验室检查不支持血管性、炎症性、感染性、中毒、肿瘤等病变,考虑神经系统变性、遗传及代谢性疾病可能,入院头颅MRI、DWI显示持续存在的皮髓质交界处高信号,结合上级医院皮肤活检发现部分汗腺细胞胞核内嗜酸性包涵体,p62和泛素免疫组织化学染色阳性,最终散发型成人型神经元核内包涵体病(NIID)诊断成立。
NIID是一种罕见的缓慢进展的神经变性病,其可累及多个部位,其中包括中枢神经系统、周围神经系统、自主神经系统以及其他器官,细胞核中可发现嗜酸性透明包涵体[1],于1968年由Lindenberg首次提出,可经皮肤活检确诊。NIID根据起病年龄分为成人型和未成年型,成人型分为散发型和家族型,未成年型根据年龄分为儿童型和青少年型[2]。散发型NIID患者发病年龄在51~76岁。散发型的首发症状多为痴呆,自主神经功能受损[3]亦常出现。本例患者与大部分病例报道相似,表现以痴呆为主的临床症状,但本例患者仅表现认知功能受影响,不合并有其他症状,如头痛、疲劳等,这在既往报道中较少见,我们猜测可能与该患者处于疾病早期相关,需后期随访、关注有无其他症状出现。
NIID的临床症状具有高度异质性,目前也没有大量的临床试验研究,且尚无特异性治疗。由于NIID发病率极低,因此临床及影像医师对其认识不够,基层医院尤甚。本文主要复习该病相关影像学表现的文献,旨在加强临床及影像医生对NIID的影像学认识,降低误诊漏诊率。
儿童以及少年起病的NIID患者影像学表现不典型,头颅MRI或CT可无明显异常[4],一些患者头颅MRI或头颅CT可出现第四脑室扩大[5],而有小脑症状表现的患者影像学多可表现为小脑萎缩[6]。一些成人起病的NIID病例磁共振FLAIR序列上可见小脑蚓部旁及小脑中脚高信号,也可见小脑萎缩,这也具有一定特征性,或可作为诊断NIID的参考指标[7]。与儿童及少年起病型相比,大多数成人NIID患者头颅MRI可表现为T2序列和FLAIR序列上的双侧对称性弥漫性的脑白质病变,DWI序列上可出现皮质髓质交界处分布的持续性异常高信号病变,此影像学表现为最具特征性的表现,国内陈为安[8]将该征象命名为皮质下绸带征(subcortical lace sign)。研究发现[9],该异常皮髓交界处的DWI序列高信号早期多出现在额叶,这或可解释多数成人患者早期以皮层症状起病,而随着患者患病时间的延长,研究发现[10]DWI序列高信号会逐渐向其他皮髓质交界区发展,但无论病情如何进展病变均不会波及深部髓质,研究显示100%的散发型与81.8%的家族型NIID均有皮质下绸带征表现,无论临床是否存在明显的认知障碍症状。除典型MRI表现外,磁共振波谱成像(MRS)可见NAA/Cr值下降、磁共振弥散张量成像(DTI)可见白质纤维束受损、正电子发射计算机断层显像(PET-CT)可见双侧大脑半球(尤其是额顶叶皮质)葡萄糖代谢减低,这些高级影像学改变可能与认知功能障碍有关[11]。研究发现,在表现为亚急性起病的以脑病症状为主的患者中,增强核磁共振可出现病灶的局灶性水肿和不同程度的病灶强化[12]。少数患者MRI上可出现类似急性缺血样的表现。本文报道的患者起病形式亦类似急性缺血样表现,因此早期导致误诊,需引起临床医师重视。
目前皮髓交界区DWI高信号的病理学机制仍不明确。Yokoi等[13]尸检了1例NIID患者,发现皮髓交界区的线样DWI高信号与近U形纤维的皮质下白质海绵状病理改变分布一致,表明此两者之间可能存在相关性。另外一例随访10年的报道显示,DWI上该病灶范围会随着病程的进展沿皮髓交界区扩展,最终表现为“水渍样”分布[14]。但近来又有报道一例62岁经皮肤活检确诊的NIID患者,其DWI高信号在5年后消失[15],该文章作者推测这种信号改变可能与随后的神经元丢失与胶质细胞增生有关。关于DWI像高信号病灶随时间演变的特点可能还需要更多的病例、更长时间的随访进一步明确。
综上,NIID影像学表现具有一定的特异性,尤其是DWI序列皮质髓质交界处分布异常高信号病变,临床及影像医师需对该影像学表现了然于胸,做到过目不忘,进一步可尽量联系皮肤活检,做到早期诊断。关于DWI像高信号病灶随时间演变的特点还需要更多的病例、更长时间的随访,且有关该病其他影像学特点也有待进一步研究。
猜你喜欢
湘潮(上半月)(2022年8期)2022-12-12
放射学实践(2021年10期)2021-10-27
国际医学放射学杂志(2020年1期)2020-02-19
影像研究与医学应用(2020年1期)2020-02-06
中国生殖健康(2020年6期)2020-02-01
中国生殖健康(2018年6期)2018-11-06
大观(2018年8期)2018-01-23
中国医药指南(2017年3期)2017-11-13
中国医学影像技术(2017年5期)2017-06-05
扬子江(2016年1期)2016-05-19
