
同种异体CAR-T细胞的研究进展
2021-08-22 01:30:34方雅玲董宇倩王亚男吴维怡
肿瘤 2021年5期
方雅玲,董宇倩,王亚男,吴维怡
嵌合抗原受体(chimeric antigen receptor,CAR)-T 细胞技术代表了癌症免疫治疗领域的重大突破。采用合成生物学的方法,免疫细胞表达经工程改造的CAR,CAR-T 细胞进入肿瘤部位,特异性识别并杀死肿瘤细胞,并进一步促进肿瘤抗原的释放,由此触发CAR-T 细胞的广泛增殖,并激活免疫系统引发进一步的抗肿瘤反应[1]。目前,已获美国食品药品监督管理局(Food and Drug Administration,FDA)批准的2 款CAR-T 细胞[美国诺华公司(Novartis)的CTL019(商品名:Kymriah®)和美国风筝制药公司(Kite Pharma)的Axi-Cel(商品名:Yescart®)]已用于治疗复发或难治性B 细胞急性淋巴母细胞性白血病,复发或难治性弥漫性大B 细胞淋巴瘤和原发纵隔大B 细胞淋巴瘤[2-3],但使用的都是自体CAR-T 细胞制备技术。自体CAR-T 细胞治疗需要为每个患者定制生产过程,生产成本因无法规模化而居高不下;生产周期较长,某些高度增殖性疾病(例如急性白血病)患者可能会错过最佳治疗窗口;有些患者自身的T 细胞存在数量缺陷或细胞功能障碍,导致CAR-T 细胞的质量得不到保证。在这种治疗背景下,同种异体CAR-T 细胞疗法应运而生。同种异体CAR-T(allogeneic CAR-T),又称通用型CAR-T(universal CAR-T)和即用型CAR-T(off-the-shelf CAR-T)是指将来源于外周血和脐带血[4],或者衍生自可再生干细胞的T 细胞[5],进行基因改造后使其既能规避移植物抗宿主和宿主排斥,又能发挥既定的抗癌作用。该免疫疗法在过去几年中的代表性里程碑事件见表1。
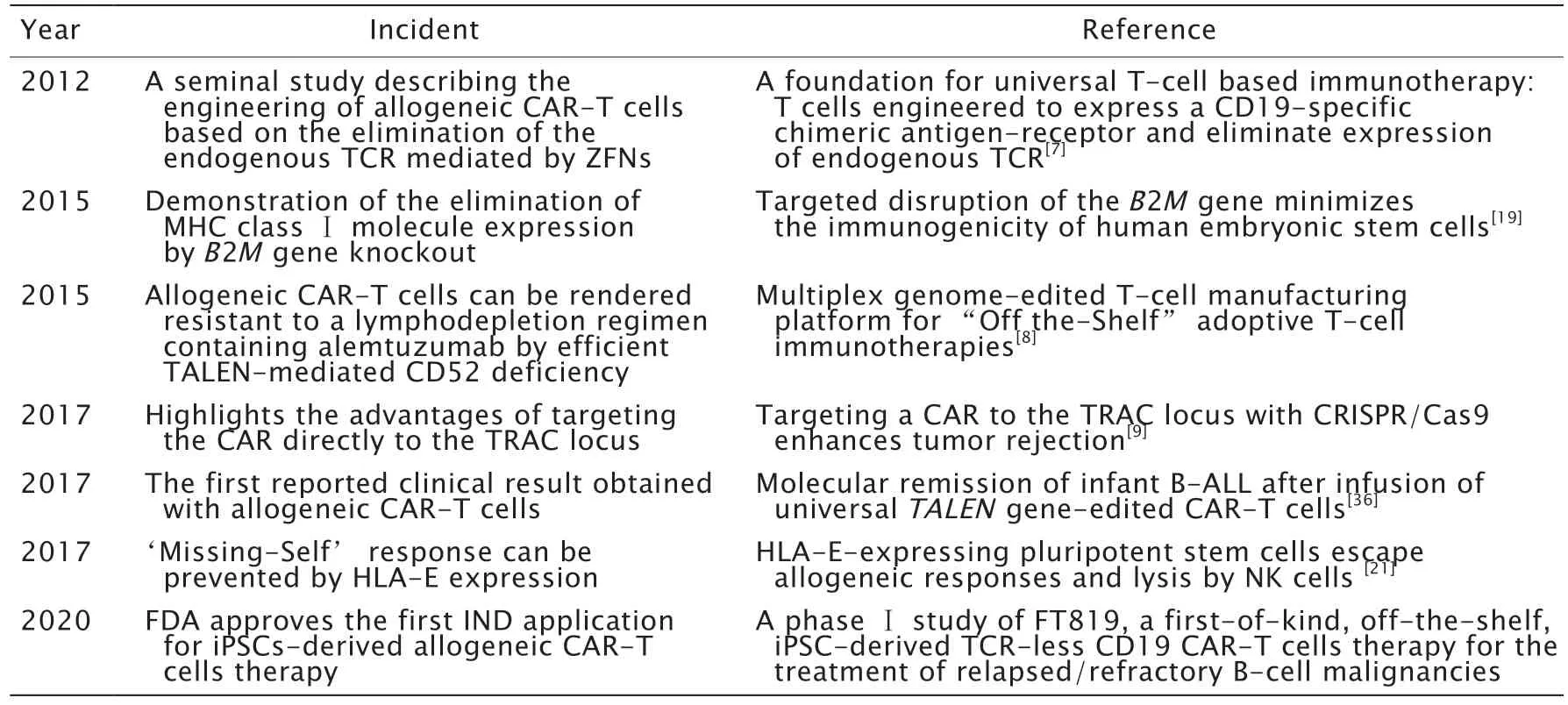
表1 同种异体CAR-T 细胞发展的里程碑事件Table 1 Milestone in the development of CAR-T cells
通用型CAR-T 细胞移植的最大障碍在于移植物抗宿主病(graft-versus-host disease)和宿主排斥异源T 细胞。一方面,输入患者体内的同种异体T 细胞上的T 细胞受体(T-cell receptor,TCR)能够识别患者的同型抗原,从而引发GvHD;另一方面,患者自身的免疫细胞可能会识别CAR-T 细胞表面上的组织相容性人白细胞抗原(human leukocyte antigen,HLA),从而攻击输入体内的CAR-T 细胞,导致免疫排斥。因此,本文将就近年来通用型CAR-T 技术在规避GvHD 和免疫排斥异源T 细胞方面的研究发展及临床前景进行综述。
1 通过基因编辑靶向敲除TCR 以规避GvHD
通用型CAR-T 技术主要依赖于基因编辑平台来解决GvHD。在基因组中特异性地造成双链缺口,然后采用非同源末端连接或同源模板重组修复,从而实现TCR 的靶向敲除并减少或消除GvHD。当前,存在3 种成熟的用于产生TCR缺陷型T 细胞的基因编辑方法:锌指核酸酶(Zinc finger nucleases,ZFNs)法、转录激活子样效应因子核酸酶(transcription activator-like effectorbased nucleases,TALENs)法和规律性间隔的短回文序列重复簇(clustered regularly interspaced short palindromic repeat,CRISPR)法。ZFNs是一类经过特殊设计的DNA 内切酶,其可以导致目标基因的双链断裂,然后通过非同源末端连接或同源重组修复断裂位点,引起核苷酸的缺失或插入,从而使靶基因功能丧失。主要功能:(1)通过改进专性异二聚体结构增强锌指核酸酶活性;(2)利用锌指酶来进行基因编辑;(3)快速构建锌指核酸酶的重组锌指阵列。TALENs 具有与ZFNs 类似的结构和功能,由特异性的DNA结合蛋白TALE 和行使DNA 切割功能的Fok Ⅰ核酸内切酶组合而成。主要功能:(1)不再像TAL-type Ⅲ effectors 一样依赖于通过结合特定的DNA 碱基组合来进行基因编辑;(2)一个简单的密码控制着TAL 效应器的识别;(3)通过TALE 核酸酶对人多能细胞进行基因编辑。基于ZFNs 和TALENs 的方法都需要为每个新的基因靶点设计特定的核酸酶对,这限制了该技术的广泛应用。CRISPR-Cas 系统是从原核生物进化获得的一套特有天然免疫系统,通过小的CRISPR RNAs 靶向抵抗病毒侵袭,如今被修饰成靶向哺乳动物细胞DNA,可实现对DNA 序列的修剪、切割、替换或添加。该技术可对人类细胞中的 RNA 程序化基因组进行编辑;使用 CRISPR/Cassystems 对多重基因组进行编辑;通过 Cas9对 RNA 引导的人类基因组进行编辑;使用 Cas9 RNA 引导的核酸内切酶在人类细胞中进行靶向基因组的编辑。该系统只需要设计一个靶向目标序列的单导核酸(single-guide RNA,sgRNA)就能引导Cas 核酸酶作用于相应的结合位点。
TCR 蛋白复合物由α 链和β 链(在αβT 细胞中)或γ 链和δ 链(在γδT 细胞中)组成,并与诸如CD3 蛋白的辅助分子相关。由于αβTCR受体需要形成异二聚体才能表达功能性细胞表面分子,因此敲除TCR 恒定α 链(TCR alpha constant,TRAC)或β 链(TCRbeta constant,TRBC)足以消除αβTCR 的表达。但是考虑到β链基因包含2 个恒定区,而编码α 链的基因只有1 个,因此最直接破坏αβTCR 的方式就是靶向编码TRAC 的基因。2011 年发表的一项研究支持了这一观点,TCR α 亚基恒定基因发生突变会导致人类免疫缺陷病其特征是αβTCR 阳性T 细胞的缺失[6]。另一项开创性研究表明,通过ZFNs可以编辑基因消除内源性αβTCR 的表达,这些重新编程的T 细胞不仅表现出对CD19 的定向特异性,不响应TCR 刺激,而且保持了应有的抗肿瘤功能[7]。此外,TALENs 亦被证明,可在原代T 细胞中实现高效的基因编辑。例如,通过电穿孔技术将TALEN mRNA 转入宿主细胞可获得αβTCR 缺陷型T 细胞,从而消除了T 细胞与同种异体抗原的反应,并介导GvHD 的可能性[8]。同样基于对TALENs 的应用,研究人员在制备用于治疗难治性或复发性CD19 阳性的B 细胞白血病的UCART19 时,敲除了TCR α基因,旨在消除供体T 细胞引发GvHD 的可能性,同时敲除了CD52 基因可使供体T 细胞对淋巴瘤抑制剂阿仑单抗产生耐药性[12]。出于安全性的考虑,UCART19 试验中还进一步通过基因工程改造共表达的RQR8 基因。激活CAR-T 细胞上所携带的RQR8 安全开关,可允许在单次高剂量给药利妥昔单抗时特异性清除异常扩增的CAR-T细胞[12]。另外,发表在Nature 上的一篇报道则突显了CRISPR/Cas9 基因组编辑促进免疫治疗的潜力[9]。研究发现,将针对CD19 特异性的CAR 导入TRAC基因座,不仅可以使T 细胞产生均匀的CAR 表达,而且还可以增强T 细胞的抗肿瘤疗效[9]。进一步研究发现,将CAR 定位到TRAC基因座还可避免强直性CAR 信号转导,并有效延缓效应T 细胞的分化和衰竭[9]。此外,megaTAL 核酸酶[10]和I-CreI 归巢核酸内切酶[11]也被开发用于靶向清除内源性TCR 的表达。以上发现不仅揭示了破坏CAR-T 细胞内源性TRAC 表达的可行性,也突显了基因组编辑TCR缺陷T 细胞以规避GvHD 的潜力。然而,需要指出的是,异体CAR-T 细胞在被敲除TCR基因后,仍然需要经历TCR 负性筛选[13]。尽管目前的筛选效率较高,但依然无法保证极少量的TCR阳性细胞不产生GvHD。这也就意味着,当前临床上应用同种异体CAR-T 细胞,仍然伴随着治疗细胞的数量与功效之间的权衡。
基因编辑正在彻底改变细胞免疫治疗的可能性,但这也带来了未知的风险。基因脱靶随时可能发生,当多个位点被切断时,新的易位可能发生。在UCART19 疗法中,可以看到CD52 和TRAC基因座之间的重组[8]。
2 提高同种异体CAR-T 细胞的持久性
虽然上述有多种方法可以降低输入到患者体内的异体CAR-T 细胞攻击宿主的风险,但是同种异体CAR-T 疗法还需要解决另一项重大挑战,那就是患者的免疫系统会识别这些细胞是“外来”细胞,从而对它们产生免疫排斥反应。这种免疫排斥可能最终会将输入患者体内的异体CAR-T细胞完全消灭。而由患者自身T 细胞介导的免疫反应可能在异体CAR-T 细胞疗法输入时就马上开始工作,降低异体CAR-T 细胞疗法的疗效。因此,如何提高同种异体CAR-T 疗法的持久性是这一领域亟待解决的问题。
2.1 淋巴细胞耗竭及其与基因编辑相结合的策略
一种增加异体CAR-T 细胞持久性的可能方案是,在不影响CAR-T 细胞活性的前提下延长淋巴细胞清除的持续时间。有研究显示,将异体CAR-T 细胞转移到淋巴细胞耗竭的人体后会经历一个称为稳态扩增的过程[15]。这一扩增是由诸如白细胞介素7(interleukin-7,IL-7)和IL-15在内的细胞因子以及暴露于靶标抗原所驱动[16]。因此,为确保CAR-T 细胞能发挥最大功效,在回輸之前,患者要先接受高剂量化疗尽可能清除体内的原有淋巴细胞(lymphodepletion)以期在患者体内构建一个更有利于CAR-T 细胞生长的免疫环境[15]。一项研究表明,使用电穿孔技术将TALEN mRNA 转入原代人T 细胞中进行高效的多重基因编辑。这种TALEN 介导的编辑方法可以使T 细胞同时缺失αβTCR 和CD52 蛋白的表达[12]。TCR 和CD52 缺陷的CD19 靶向型CAR-T 细胞可以在阿仑珠单抗清除体内淋巴细胞时稳态扩增[12]。在淋巴瘤原位小鼠模型中,UCART19 的抗肿瘤活性与标准的CD19 靶向性CAR-T 细胞相比没有区别[8];且UCART19 疗法在临床上的初步试验也进一步支持了这一方案的可行性[17]。这项UCART19 疗法治疗高风险难治性或复发性B 细胞白血病患者试验的初步结果显示,UCART19 治疗没有带来意外的毒性,且在CAR-T 细胞输注后的第42 天依然可以在患者的血液中检测到,证实其在人体内的持久扩增能力[17]。但是,在采用这一策略时患者体内的内源 细胞水平会被压制到很低的水平,感染风险显著提高。
2.2 通过基因编辑规避异源细胞的免疫原性
同种异体CAR-T 细胞的治疗窗口取决于其初始扩增,持久性以及宿主免疫系统对它们的排斥程度。目前已有诸多研究人员对来自具有不同HLA 表达供体的T 细胞进行基因编辑,以逃避免疫应答。由于HLA-I 型蛋白(包括HLA-A、HLA-B 和HLA-C 等)是介导免疫排斥因素的关键分子,通过基因工程改造的方法敲除同种异体CAR-T 细胞表达HLA-I 型蛋白将最为直接。有研究针对HLA-A 进行了特定的ZFN 核酸酶设计,电转编码这些工程核酸酶的mRNA 选择性破坏CD19 特异性T 细胞上HLA-A 的表达,最后成功对HLA-A 阴性T 细胞通过HLA 限制性细胞毒性T 细胞进行了富集[18]。
HLA 在细胞表面表达时需要β2-微球蛋白(β2-microglobulin,B2M)的亚基充当HLA-Ⅰ轻链,所以通过基因工程敲除B2M基因,也可以导致细胞表面缺乏功能性HLA-Ⅰ的表达。一项研究报告了新型B2M(-/-)人类胚胎干细胞(humanembryonic stem cells,hESCs)系的产生。从该系分化出的成熟T 细胞即使在干扰素γ(interferon-γ,IFN-γ)的刺激下也不表达细胞表面HLA 分子,免疫原性远低于正常hESCs[19]。此外,B2M(-/-)hESCs 品系不发生脱靶整合或切割事件,没有稳定的B2M mRNA,并保留了自我更新能力,基因组稳定性和多能性[19]。使用CRIPSR-Cas9 系统,研究人员对T 细胞的内源性TRAC、B2M和程序性死亡受体1(programmed cell death protein 1,PD-1)进行多重基因破坏,创建了可以抵抗PD-1 抑制的通用性CAR-T 细胞三联基因编辑的CAR-T 细胞在临床前的神经胶质瘤模型中显示出显著增强的抗肿瘤活性,且模型小鼠接受该CAR-T 细胞治疗后,显著延长存活时间[20]。
通过基因工程手段使T 细胞表面缺失HLA的表达,不可避免的就是对宿主自然杀伤(natural killer,NK)细胞的清除更加敏感。针对这个问题,已开发出相应的策略。研究人员使用腺相关病毒(adeno-associated virus,AAV)介导的基因编辑,使细胞表面表达HLA-E 单链二聚体(与B2M 融合)或三聚体(与B2M 和肽抗原融合),无细胞表面HLA-A、HLA-B 或HLA-C 表达的方式,在人类多能干细胞中的B2M基因座处插入HLA-E基因。这些经HLA 改造的多能干细胞及其分化衍生物将不被CD8+T 细胞识别为同种异体,不结合抗HLA 抗体,并且对NK 细胞介导的清除具有抗性[21]。此外,还有研究人员通过CRISPR-Cas9 靶向破坏HLA-A 和HLA-B,但保留HLA-C 来增强诱导多能干细(induced pluripotent stem cells,iPSCs)的免疫相容性,逃避CD8+T 细胞和NK 细胞的杀伤作用[22]。
3 临床前景
同种异体CAR-T 细胞的诸多优势为治疗复发性或难治性恶性肿瘤提供了更多可能性,尤其在疾病进展迅速的急性髓细胞性白血病(acute myeloid leukemia,AML)和急性淋巴细胞白血病(acute lymphocytic leukemia,ALL)中有主要优势。突破性的实例如,2例患有顽固性难治性CD19 阳性的B 细胞ALL 的婴儿在接受单剂量UCART19 细胞(4.0~4.6×106个/kg)输注后,均在28 d 内达到了分子缓解,并且该UCART19细胞在婴儿体内一直持续存在,直到成功完成同种异体干细胞移植。这种移植前的过渡性策略证明了基因编辑技术的治疗潜力。目前有多项同种异体CAR-T 细胞治疗AML 和ALL 的临床试验正在开展,亦有试验针对淋巴瘤、多发性骨髓瘤和实体瘤(表2)。所涉及的靶标与自体CAR-T治疗的靶标相似,包括ALL 和B 细胞淋巴瘤中的CD19 和CD22;多发性骨髓瘤中的BCMA和CS1(也 称CD319 和SLAMF7);AML 中的CD123;T 细胞白血病和T 细胞淋巴瘤中的CD7 以及实体瘤中的NKG2D(natural killer group 2 member D)、CD70 和间皮素等。值得注意的是,对于AML 中的CD123 和多发性骨髓瘤中的CS1 等靶标,它们在肿瘤细胞上异质表达或在正常造血祖细胞上表达[23],脱靶效应会是CAR-T 细胞不良反应的主要来源。但同时,由于同种异体CAR-T 细胞的持久性有限,对于靶点在正常细胞上表达,异源性CAR-T 细胞的较短的持久性可能会变得有利。此外,将不同特异性的异源CAR-T 细胞组合在一起,以消除肿瘤异质性的影响或避免初始靶标丢失的免疫逃逸,或许在将来会是一种有吸引力临床治疗策略。例如,在ALL 或B 细胞淋巴瘤中使用CD19 和CD22的CAR-T 细胞的组合[24]。近期,美国FDA 已经批准首个iPSCs 来源的同种异体CAR-T 细胞疗法FT819 的新药研究申请(Investigational New Drug),用于治疗复发/难治性B 细胞恶性肿瘤,包括慢性淋巴细胞白血病、ALL 和非霍奇金淋巴瘤等。FT819 目前处于Ⅰ期研究阶段,这是一种首创的、现成的、由iPSC 衍生的少TCR的CAR-T细胞疗法,用于治疗复发性/难治性B 细胞恶性肿瘤。iPSCs或将成为推进免疫细胞疗法产业化的重要支柱。
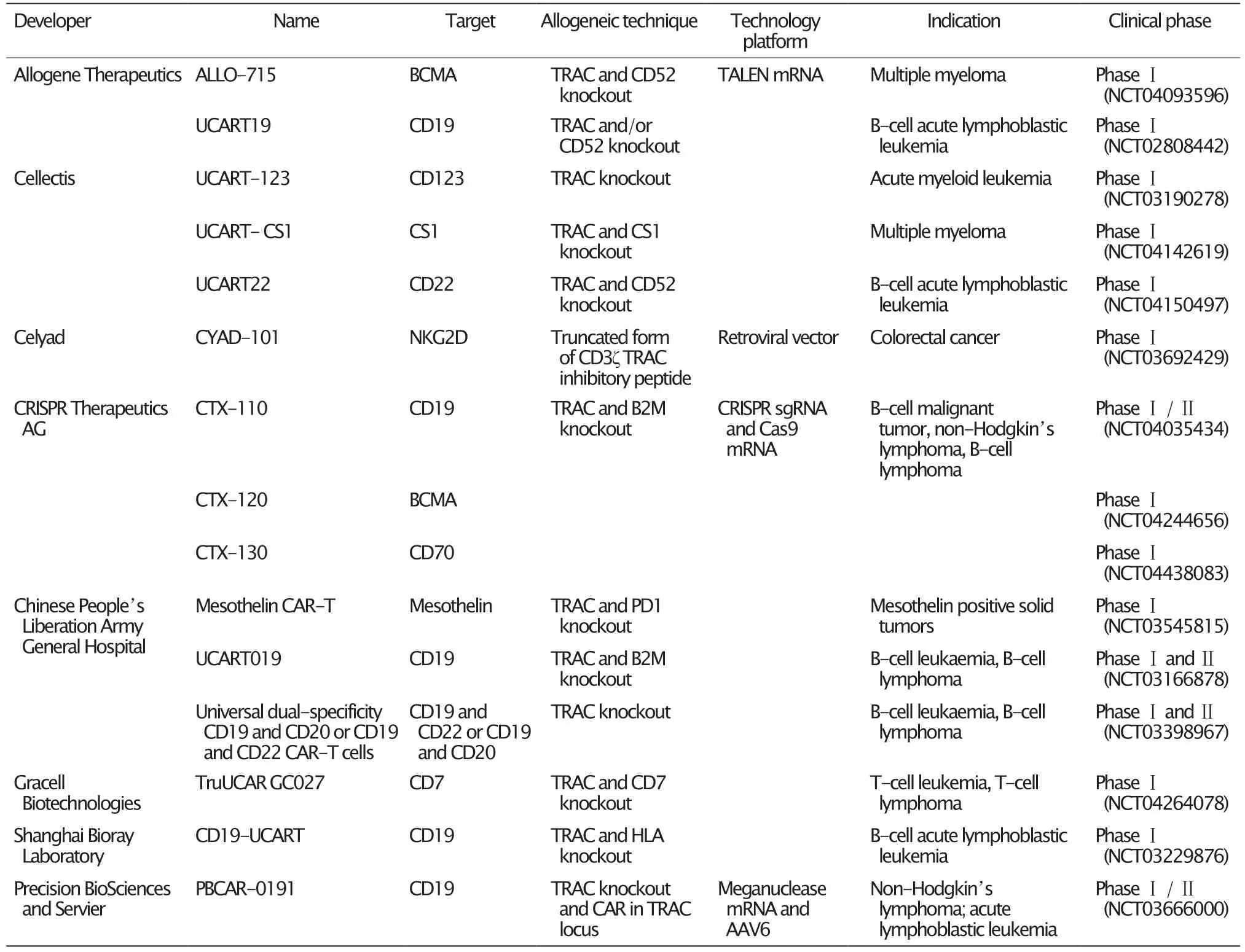
表2 进入临床开发阶段的代表性通用型CAR-T 细胞产品Table 2 Representative universal CAR-T cells products entering the clinical development stage
对于面临多重挑战的实体瘤,一方面,肿瘤微环境中的物理屏障让CAR-T 细胞难以浸润到肿瘤内部;另一方面,即使CAR-T 细胞浸润到了实体瘤内部,也面临着肿瘤微环境的免疫抑制[25]。目前,已经有许多功能性模块用于CAR-T 细胞的工程化改造,这些模块包括改善T 细胞归巢和运输的C-C 基序趋化因子受体29(C-C motif chemikon receptor 29,CCR29)、CCR4 和CCR1[27-28];改 善T 细胞增殖和持续性的细胞因子IL-12[29]、IL-18[30]、IL-15 和IL-21[31]分泌元件和细胞因子IL-7受体(IL-7 receptor,IL-7R)分泌[32];对抗免疫抑制的显性失活的(dominant-negative)受体[33]和开关受体[34];局部呈递免疫检查点阻断剂的元件[35]等。因此,目前有诸多策略来优化下一代CAR-T 细胞,使其具有更好的肿瘤选择性、更好的肿瘤浸润能力和在肿瘤微环境中对抗免疫抑制的能力。基因编辑技术允许在同种异体的CAR-T细胞中组合多种修饰,然而它们必须满足严格的质量控制和监管认证流程,以应对可能的临床风险。
4 展 望
CAR-T 细胞疗法已经改变了某些血液系统恶性肿瘤的治疗模式,并且是癌症治疗邻域最有希望的疗法之一。自体CAR-T 细胞治疗正在集中阐述肿瘤不完全消除的机制,研究如何降低毒性,防止抗原逃逸以及基于已建立的合成生物学原理确定合适的靶标和策略,并将该方法扩展到其他恶性肿瘤。通用型CAR-T 细胞治疗除了面临着同样的挑战,还面临着GvHD 和异体排斥这两大难题。此外,同种异体CAR-T 细胞的简化生产亦值得深入研究。例如直接生成超纯现成同种异体 CAR-T 细胞a。但有理由相信,随着基因编辑技术的发展,现货型细胞治疗将成为未来细胞疗法领域的重要方向,有望为更多患者提供可获得的治疗途径。
猜你喜欢
北方牧业(2023年13期)2023-07-28 06:50:54
保健医苑(2022年5期)2022-06-10 07:46:38
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
无机化学学报(2020年7期)2020-07-20 02:06:44
三农资讯半月报(2020年8期)2020-05-13 14:26:35
汉字汉语研究(2019年2期)2019-08-27 00:47:52
中国篆刻(2017年7期)2017-09-05 10:01:36
中国篆刻(2017年8期)2017-09-05 09:44:30
中国篆刻(2017年5期)2017-07-18 11:09:31
