
铁死亡在急性肾损伤和肾癌等相关疾病中的研究进展
2021-08-18 09:08叶承林综述刘修恒审校
疑难病杂志 2021年8期
叶承林综述 刘修恒审校
细胞死亡有多种形式,在20世纪60年代,人们认识到细胞死亡可以通过分子机制来调控,并且可以为正常生理功能提供帮助,也能导致病理变化,“程序性细胞死亡”这一概念随之被提出。细胞死亡曾被分为三大类,Ⅰ型细胞死亡即细胞凋亡;Ⅱ型细胞死亡又叫自噬相关性细胞死亡;Ⅲ型细胞死亡被称为细胞坏死[1]。最近的研究发现,调节性细胞死亡有多种形式,其分子机制和形态特征各不相同。铁死亡(ferroptosis)这一概念于2012年首次提出,是一种铁依赖的非凋亡性细胞死亡。这种细胞死亡机制不需要含半胱氨酸的天冬氨酸蛋白水解酶(caspase)的激活,也不需要其他凋亡效应分子(如BAX或BAK)的参与,也不伴随凋亡的形态特征或生化过程。形态学上主要表现为线粒体的缩小、线粒体嵴减少或消失[2]。研究证明铁死亡在肿瘤、缺血再灌注、神经退行性病变等领域发挥重要作用,其在肾脏相关疾病中的重要性也逐渐被发现,本文就铁死亡的发现、作用机制与调控及在肾脏疾病发生发展中的作用展开综述,为肾脏疾病的进一步研究提供新的方向。
1 铁死亡概念
在铁死亡的概念被提出及详细分子机制发现之前,已经有很多文献报道了对于这一现象的观察,但都被归因于其他的细胞死亡机制,或者被认为没有生物学意义。20世纪50年代中期Eagle等[3-5]发现13种不同氨基酸中只有一种氨基酸饥饿抑制了人和小鼠细胞的生长;被剥夺胱氨酸的细胞表现出独特的显微形态,与被剥夺其他氨基酸时的形态不同,推测与病毒感染引起的细胞死亡相似。1977年Bannai等[6]发现胱氨酸饥饿导致细胞谷胱甘肽减少和细胞死亡,通过添加亲脂性抗氧化剂α-生育酚(α-tocopherol,维生素E的一种成分)可以挽救这种胱氨酸剥夺所致的死亡,从而支持了活性氧(ROS)积累在细胞死亡诱导中的贡献,并且这一过程无需谷胱甘肽参与。Ratan等[7]发现由于剥夺血清而导致的铁缺乏可以阻止细胞胱氨酸剥夺和谷胱甘肽耗竭在诱导细胞死亡中的关键作用,现在被认为是铁死亡的特征表现之一。
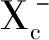
Dixon等[11]于2012年发现首个铁死亡抑制剂ferrostatin-1,并且在这些研究基础上正式提出了铁死亡(ferroptosis)这一概念,用以描述这种铁依赖性非凋亡性的细胞死亡。
2 铁死亡机制与调控

2.2 脂质过氧化与活性氧(ROS)的蓄积 ROS通常被认为是氧消耗和细胞代谢的副产品,由分子氧的部分还原形成[21]。肿瘤内源性活性氧主要来源于细胞线粒体及NADPH氧化酶(NOXs)[22]。过量的ROS被抗氧化剂通过酶促或非酶促反应及在超氧化物歧化酶(superoxide dismutase,SOD)如Cu/Zn-SOD、Mn-SOD等,谷胱甘肽过氧化物酶(glutathione peroxidase,GPX)和过氧化氢酶催化的反应中解毒。ROS产生和解毒速率的不平衡会导致氧化应激,并由此产生自由基,这些自由基会破坏DNA、蛋白质和脂质。氧化还原活性金属,特别是铁,可以通过芬顿反应促进细胞中的ROS聚集。在铁死亡过程中,胱氨酸—谷氨酸逆向转运蛋白(recombinant solute carrier family 7,member 11,SLC7A11)的失活和谷胱甘肽的耗竭导致铁依赖的ROS积聚[15]。
多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)的过氧化是铁死亡的主要驱动因素。铁依赖的脂质活性氧的积累和随后多不饱和脂肪酸磷脂(PUFA-PLS)的消耗是铁死亡的共同特征[23]。花生四烯酸(arachidonic acid,AA)是目前已知的铁死亡细胞中最常消耗的多不饱和脂肪酸[24]。Dixon等[25]的研究发现,将花生四烯酸插入膜磷脂中所涉及的关键酶缺失可以防止铁死亡的发生。酰基辅酶A合成酶长链家族成员4(acyl-CoA synthetase long chain family member 4,ACSL4)可通过促进可氧化的细胞膜磷脂的积累来驱动铁死亡[26]。

表1 铁死亡调控相关诱导剂与抑制剂
3 铁死亡与急性肾损伤
急性肾损伤(acute kidney injury,AKI),也称为急性肾衰竭(acute renal failure,ARF),是由多种原因引起的常见严重疾病,病因包括肾缺血、肾毒性药物和尿路阻塞[32]。AKI的发病机制非常复杂,由于其解剖学特征和复杂功能,肾单位的近端肾小管节段最容易受到各种形式的损伤[33]。已经发现了多种分子机制可以诱导或加重AKI,其中ROS被认为是诱导肾损害的关键介质之一[34]。多项研究表明,铁死亡是潜在的治疗靶点,尤其是在肾小管坏死占主导的疾病中[35]。
在肾脏中,缺血再灌注损伤(ischemia-reperfusion injury,IRI)是AKI的主要原因之一。IRI的特征是特定器官的血液供应突然停止并在血流恢复后重新供氧。该过程可通过触发涉及ROS、细胞因子、趋化因子和白细胞激活的炎性级联反应来加剧组织损伤[36-37]。以往在各种缺血性损伤模型中,凋亡被认为是主要的调控性细胞死亡。当坏死性凋亡被发现时,这种受体相互作用的丝氨酸/苏氨酸激酶3(receptor interacting serine/threonine kinase 3,RIPK3)和混合谱系激酶结构域样(mixed lineage kinase domain like,MLKL)依赖性调节性坏死的形式被认为是心脏及肾脏缺血性损伤的主要原因[38-39]。最近的发现表明,铁死亡可能是缺血性损伤的主要驱动力之一。在严重的IRI模型中应用铁抑素(ferrostatin)可保护小鼠免受功能性急性肾衰竭,然而RIPK1抑制剂necrostatin-1(Nec-1)不能保护新鲜分离的肾小管免受缺氧损伤[40],这突显了铁稳态在人类缺血再灌注损伤中的重要性。基础研究证明,NRF2过度激活会增加GSH的产生并阻止肾脏IRI早期阶段的进展[41],许多负责阻止脂质过氧化进而促进铁死亡的蛋白质和酶都是NRF2的靶基因,如SLC40A1、谷胱甘肽合成酶、GPX4。NRF2或其靶基因的功能失调与对细胞应激源的反应性下降有关,并诱导细胞铁死亡[42]。Lee等[43]研究表明,用能量应激诱导剂AICAR或2-Deoxy-D-glucose(2DG)处理可以通过抑制铁死亡保护小鼠免受肾脏IRI的影响。
横纹肌溶解引起的肾衰竭占所有急性肾衰竭病例的15%[44],研究表明,肌红蛋白(myoglobin,Mb)在肾脏中的积累是导致肾脏损害的核心机制。近年来,对横纹肌溶解诱导的急性肾损伤研究表明,Mb代谢产生的Fe2+直接诱导近端肾小管上皮细胞脂质过氧化可能是横纹肌溶解致肾损伤的重要机制之一[45]。肾脏中Mb降解释放的游离铁通过芬顿反应的催化作用参与氧化性物质的产生。研究表明,使用铁络合剂去铁胺可以减轻横纹肌溶解诱导的大鼠肾脏损伤[46]。Guerrero-Hue等[47]证明了铁死亡在横纹肌溶解诱导AKI中的关键作用,并表明铁死亡敏感的这一过程可以被强抗氧化剂姜黄素所抑制。
4 铁死亡与肾癌
肾细胞癌根据病理特征可分为肾透明细胞癌、乳头状肾细胞癌、嫌色细胞性肾细胞癌及Bellini集合管癌等,其中以肾透明细胞癌最为常见。肾癌多对化疗和放疗耐药。激活调控性细胞死亡是肾癌的理想治疗策略,并可能有助于解决肾癌的耐药性。
铁死亡首先在非小细胞肺癌HT-1080细胞系中被鉴定[11],随后,Yang等[12]测试了117个肿瘤细胞系对erastin引起铁死亡的敏感性,并发现弥漫大B细胞淋巴瘤和肾细胞癌对GPX4调节的铁死亡尤为敏感。Miess等[48]发现siRNA沉默谷胱甘肽过氧化物酶GPX3和GPX4对肾透明细胞癌细胞是致命的,并且阐述了通过抑制GSH的合成,肾透明细胞癌对于铁死亡变得更加敏感,从而抑制肿瘤的生长。在缺乏VHL基因的肾癌细胞中,重新表达的VHL产生了对铁死亡的抵抗力。最近的一项研究表明,能量应激(energy stress)介导的AMPK激活能够抑制铁死亡,在肾癌Caki-1细胞系(一种基础AMPK活性较低的铁死亡敏感细胞系)中过表达 AMPK可适度增加乙酰辅酶A羧化酶(acetyl CoA carboxylase,ACC)的磷酸化,并保护细胞免受Erastin诱导的铁死亡影响[43]。Yang等[49]的一项研究表明,Hippo通路效应器TAZ可以调节肾癌对铁死亡的敏感性。Kerins等[50]发现,延胡索酸水合酶的失活可以导致细胞内延胡索酸的蓄积从而提高遗传性平滑肌瘤病和肾细胞癌(hereditary leiomyomatosis and renal cell cancer,HLRCC)对铁死亡的敏感性。
5 铁死亡相关研究的展望
铁死亡作为一种调控性的细胞死亡,在肾脏疾病中具有双重作用,对于铁死亡在肾脏疾病中的有关机制仍需要进一步探索。铁死亡机制层面上,尽管已经发现了脂质过氧化物在诱导铁死亡中的重要作用,但仍然没有确凿的证据表明此类ROS是执行铁死亡的最下游因素,关于铁死亡的最终执行分子问题仍有待解决。铁死亡与其他调控性细胞死亡如自噬、凋亡之间的联系需要进一步深入研究。铁死亡作为细胞调控性死亡的一种形式,在其作用过程中不可逆点的位置,正在进行铁死亡这一过程的细胞在何时可以被挽救,这些都需要深入的研究。铁死亡是肾脏疾病中非常重要的细胞死亡形式之一,随着相关研究的不断进展,一定会为肾脏疾病的治疗带来新的方向。
猜你喜欢
长春中医药大学学报(2022年5期)2022-05-24
现代养生·上半月(2021年10期)2021-09-24
昆明医科大学学报(2021年1期)2021-02-07
家庭科学·新健康(2020年6期)2020-07-06
科学导报(2019年45期)2019-09-23
保健与生活(2019年23期)2019-09-10
家庭科学·新健康(2019年4期)2019-05-05
分析化学(2018年4期)2018-11-02
中外医疗(2017年24期)2017-11-15
莫愁·时代人物(2012年4期)2012-09-22
