
抑制剂8Q9和8QC与bromodomain结构域蛋白4作用机理的分子动力学研究
2021-08-16 07:26吴世亮孙海波尹妍妍李洪云王立飞
原子与分子物理学报 2021年3期
吴世亮,孙海波,尹妍妍,李洪云,王 青,王立飞
(山东交通学院理学院,济南 250357)
1 引 言
组蛋白乙酰化主要由所谓的“书写者”,“阅读者”和“擦除者”进行调节,bromodomain结构域(Bromodomain,BRD)是组蛋白乙酰化的“阅读者”[1].迄今为止,人类已识别出61种BRDS蛋白结构域,由约140个氨基酸残基组成.这些结构域存在于46种不同的蛋白质中,并分为8个不同的家族[2,3].BRD具有高度保守的折叠模式,主要由四个反向平行的α-螺旋(αA,αB,αC,αZ)和两个柔性环(ZA环和BC环)构成[4,5].Bromodomain结构域和末端结构域(bromodomain and extra-terminal domain,BET)家族是八个主要BRD家族的重要组成部分,包含BRD2,BRD3,BRD4和BRDT[6]四个成员.
BRD4是BET家族中重要的功能蛋白.研究表明,BRD4通过招募正向转录延长因子b(positive transcriptional elongation factor b,P-TEFb)调节靶基因的表达,在调节细胞基因转录、细胞周期、炎症等生物过程中发挥重要作用[7-10].实际上,肺癌、乳腺癌和肝癌等各种恶性肿瘤的发生和发展与BRD4的表达失调或功能异常密切相关[11-13].此外,BRD4对急性骨髓性白血病细胞的分化诱导产生重要影响[14].因此,BRD4已成为用于临床治疗各种恶性肿瘤、急性骨髓性白血病、炎性疾病等药物设计的重要靶点之一.
自2010年首次公开了有效的BET家族抑制剂JQ1以来[4],许多针对BET家族特别是靶向BRD4的小分子抑制剂被广泛报道,如IBET151[15],I-BET762[16]和RVX-208[17-18]等,这些抑制剂可以诱导细胞周期停滞和凋亡,并抑制肿瘤的侵袭和转移.然而,抑制剂与BRD4蛋白的结合方式以及蛋白构象变化的细节仍然不足,这限制了针对BRD4的有效抑制剂的设计与开发.因此,进一步从原子层次了解抑制剂和BRD4的结合机制变得至关重要.
随着计算方法的发展,分子动力学模拟[19,20]和结合自由能计算[21,22]已成为探讨抑制剂与蛋白质结合机制的有效工具.在这项工作中,选择了两种抑制剂8Q9和8QC来阐明抑制剂与BRD4(1)的结合机制[23],我们之所以选择这两个小分子抑制剂是因为它们显示出与BRD4(1)不同的结合能力,对应于8Q9和8QC的IC50值分别为2270和150 nM.图1展示了抑制剂8Q9、8QC以及抑制剂/BRD4(1)复合体的结构.抑制剂8Q9和8QC与BRD4(1)的结合差异的阐明对于理解抑制剂与BRD4的结合机理具有重要意义.因此,本工作采用分子动力学模拟,结合自由能计算和抑制剂-残基相互作用来研究两种抑制剂与BRD4(1)的结合方式以及抑制剂结合导致的BRD4(1)结构的变化.同时,我们也希望这项研究能够为设计有效抑制BRD4(1)活性的抑制剂提供有价值的信息和理论指导.

图1 分子的结构:(a)抑制剂8Q9与BRD4(1)复合物的结构,(b)抑制剂8Q9的结构,(c)抑制剂8QC的结构.Fig.1 Structures of molecules:(a)the8Q9 and BRD4(1)complex,(b)the inhibitor 8Q9,(c)the inhibitor 8QC.
2 理论和方法
2.1 模拟系统的准备
动力学模拟的初始模型取自蛋白质库中的晶体结构5Y93和5Y94[23],5Y93是抑制剂8Q9和BRD4(1)的复合物(8Q9/BRD4(1)),5Y94是抑制剂8QC和BRD4(1)的复合物(8QC/BRD4(1)),apo/BRD4(1)表示从晶体结构5Y94中去掉抑制剂8QC获得的BRD4(1)的无抑制剂状态,该结构用于比较抑制剂结合对BRD4(1)构象变化的影响.所有结晶水分子都保留在初始模型中,借助Amber18中的LEAP模块,将晶体结构中缺失的氢原子连接到相应的重原子.对两种小分子抑制剂,使用了Amber18的antechamber程序中的AM1方法来实现结构优化和计算,并将原子的BCC电荷分配给抑制剂的各原子.蛋白质和水分子的力场参数由Amber18中的ff14SB力场产生,而小分子抑制剂的力场参数由GAFF力场产生.复合体被溶解在显性的TIP3P水盒子里,水盒子边缘与复合体最近原子的距离设定为12.0Å,然后将三个氯离子置于水和复合体组成的系统中,以确保系统呈电中性.
2.2 分子动力学模拟
对于每个复合体系统,通过Amber18中的PMEMD模块执行了能量最小化和分子动力学模拟.为了消除一些原子间的不合理接触,每个系统都经过了两阶段的能量优化.首先,对溶质进行约束并优化溶剂.其次,整个系统实现了无约束的优化.在优化过程中,首先执行2500步的最陡下降优化,接着进行2500步的共轭梯度优化.之后,将每个系统在2 ns内保持恒定体积逐渐从0 K加热到300 K,接着在常温300 K,常压1标准大气压条件下进行1 ns分子动力学平衡.最后,进行200 ns的无约束分子动力学模拟.在模拟过程中,每2 ps记录一次原子坐标.使用SHAKE算法来限制所有涉及氢原子的化学键的伸缩,模拟积分步长为2 fs.借助Langevin恒温器控制每个系统的温度.PME方法用于计算原子之间的长程静电相互作用.涉及静电相互作用和范德华相互作用的非成键相互作用计算的截断值设置为10.0Å.
2.3 结合自由能的计算
MM-GBSA方法是计算结合自由能的一种常用方法.在我们的研究中,借助Amber18中的MM-GBSA方法有效地估计了抑制剂与BRD4(1)的结合自由能.对于每个系统,从动力学模拟轨迹的最后80 ns中每隔400 ps取一个构象,共取出200个构象用于MM-GBSA方法的计算.计算过程中去掉构象中的水分子和离子,采用下面的公式计算抑制剂和蛋白质的结合自由能:

式中△Eele和△Evdw分别表示气相中抑制剂和蛋白质的静电相互作用与范德华相互作用,这两项用分子力学计算,力场参数由Amber18中的ff14SB力场产生;第三项△Gpol表示极性溶解自由能对抑制剂和蛋白质结合的贡献,通常由Amber套件中MM-GBSA模块求得;第四项△Gnonpol表示非极性溶解自由能对抑制剂和蛋白质结合的贡献,使用下面的方程求解:

式中SASA是溶剂可及表面积,经验常数γ和β的值分别设置为0.0072 kcal·mol-1·Å-2和0.0 kcal·mol-1;最后一项-T△S表示熵变对抑制剂和蛋白质结合的贡献,是平动,转动和振动熵之和,该项使用振动模和传统的热力学计算.
2.4 动态相关性分析
为了研究抑制剂结合引起的BRD4(1)的运动模式的改变,采用Amber 18中的CPPTRAJ程序,基于分子动力学模拟的平衡部分,对三个系统进行动态相关性分析.由第i个Cα原子和第j个Cα原子构成的矩阵元素Cij定义如下:

其中△ri表示第i个Cα原子相对于其平均值的位置偏差.Cij的取值范围是-1至+1,其中-1表示完全负相关运动,而+1表示完全正相关运动.正相关说明残基沿相同方向移动,而负相关说明残基沿相反方向移动.使用颜色编码模式来体现残基之间相关运动的程度.文中出现的DCCM图是使用分子动力学模拟轨迹的最后80 ns中保存的Cα原子的坐标计算得到的.
3 结果和讨论
3.1 分子动力学平衡
为了评估动力学平衡的稳定性,分析了三个系统的BRD4(1)主链原子相对于其初始优化结构的均方根偏差(RMSD)随时间的演化情况(图2).如图2所示,三个系统的RMSD波动不同,apo/BRD4(1),8Q9/BRD4(1),8QC/BRD4(1)达到稳定平衡的时间分别是模拟开始后10 ns,50 ns和120 ns,其平衡后的平均RMSD值分别为1.76,0.98和1.18Å,三个系统的RMSD的涨落范围均小于0.65Å,该结果说明分子动力学平衡的稳定性是可靠的,可用于随后分析的统计取样.此外,8Q9/BRD4(1),8QC/BRD4(1)两个复合体的RMSD值和涨落范围明显低于apo/BRD4(1)系统,表明抑制剂结合对BRD4(1)的结构性波动产生某些限制,这有助于抑制剂/BRD4(1)复合体的动力学稳定性.

图2 分子动力学模拟中BRD4(1)主链原子的均方根偏差Fig.2 Root-mean-square deviations(RMSDs)of the backbone atoms in BRD4(1)relative to the corresponding crystal structures as function of simulations time.
3.2 结构变化
残基的Cα原子均方根涨落(Root-meansquare fluctuations,RMSFs)可用于评估抑制剂结合对BRD4(1)结构柔性的影响.采用Amber18中的CPPTRAJ工具计算了apo/BRD4(1),8Q9/BRD4(1),8QC/BRD4(1)三个系统中各残基Cα原子涨落(图3).图3表明抑制剂的存在对BRD4(1)的结构柔性产生了重要影响.抑制剂结合的BRD4(1)中残基89-97和136-145两个区域的RMSF值比apo/BRD4(1)的要小,这表明这两个区域的柔性由于抑制剂的结合而减弱.这些结果表明,上述区域中的一些关键残基可能与两种抑制剂发生强烈的相互作用,这可以通过后面基于残基的自由能分解和氢键分析的计算进一步证明.

图3 动力学模拟过程中三个系统的BRD4(1)残基Cα原子的平均涨落Fig.3 Root-mean-square fluctuations(RMSFs)of Cα atoms in BRD4(1)of three simulated systems
3.3 动态相关性分析
为了研究抑制剂结合引起的BRD4(1)的运动模式的改变,采用Amber 18中的CPPTRAJ程序,基于分子动力学模拟的平衡部分,计算了残基之间的DCCM(图4),图中借助颜色编码模式来体现特定残基之间相关运动的程度,对角区域对应于残基相对于它们自身的运动,而非对角区域代表不同残基之间的相对运动,红色和黄色表示残基之间的强烈正相关运动,而蓝色和深蓝色表示残基之间的强烈负相关运动.
apo/BRD4(1)的DCCM如图4(a)所示,从图中可以看出在区域R1和R3(黄色)中发生了强烈的正相关运动,区域R1中的运动指示残基126-140相对于残基99-108的相关运动,而在R3区域中的运动描述残基116-121和54-59之间的相关运动;在区域R2(绿色)中注意到了一些轻微的正相关运动,描述了残基81-89相对于残基102-110的弱相关运动.与apo/BRD4(1)相比,8Q9/BRD4(1)中抑制剂的存在增强了R1和R2区域中发生的正相关运动,而大大减弱了R3区域的正相关运动,此外,8Q9的结合明显增强了区域R4中的正相关运动(图4(b)).图4(c)表明8QC/BRD4(1)中抑制剂的存在减弱了R1和R2区域中发生的正相关运动,而增强了R3区域的正相关运动,此外,8QC的结合明显减弱了区域R5中的负相关运动.以上分析表明,抑制剂的结合会导致BRD4(1)的运动模式发生重大变化,进而可能会对抑制剂与BRD4(1)中各个分离残基的相互作用产生重大影响.

图4 动态相关性分析:(a)apo/BRD4(1),(b)8Q9/BRD4(1),(c)8QC/BRD4(1).Fig.4 Dynamic cross-correlation maps(DCCMs):(a)apo/BRD4(1),(b)8Q9/BRD4(1),(c)8QC/BRD4(1).
3.4 结合自由能和作用机理
为揭示影响抑制剂结合的主要因素,比较了两种抑制剂和BRD4(1)的结合亲和力的差异.在我们的研究中,通过使用MM-GBSA方法有效地估计了两种抑制剂对BRD4(1)的结合自由能.对于每个结合抑制剂的系统,从记录在最后80 ns的分子动力学轨迹中以400 ps的时间间隔提取了200个构象用于计算.表1中的结果显示计算的结合自由能的趋势与实验值的趋势一致,这表明我们目前的自由能分析是可靠的.如表1所示,结合自由能分为五个部分,即范德华相互作用(△Evdw),静电相互作用(△Eele),极性溶解自由能(△Gpol),非极性溶解自由能(△Gnopol)和熵变贡献(-T△S).
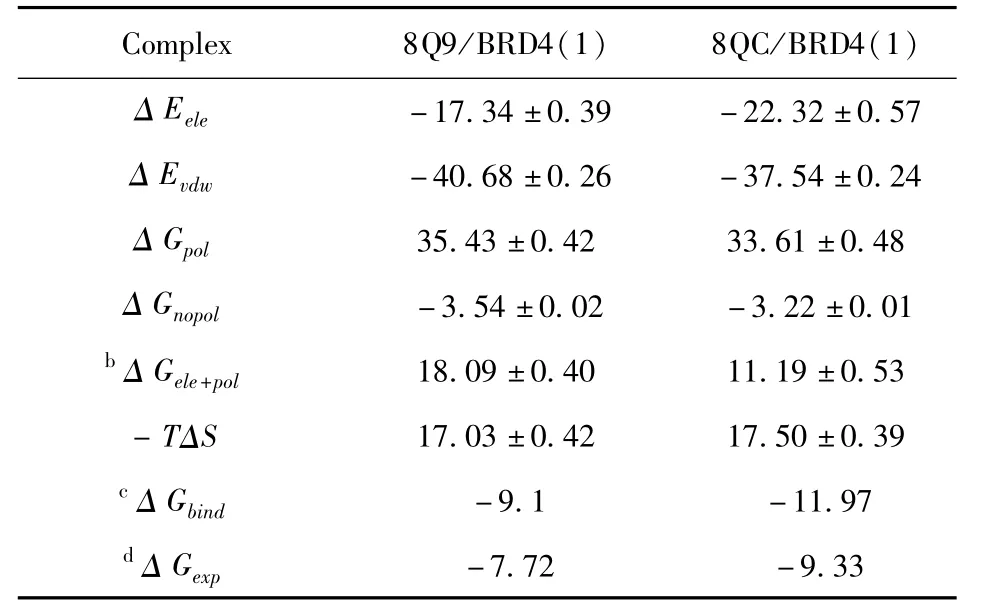
表1 MM-GBSA方法计算的抑制剂与BRD4(1)的结合自由能Table 1 Binding free energies of inhibitors to BRD4(1)calculated by MM-GBSA methoda
表1表明范德华相互作用,静电相互作用和非极性溶解自由能有利于抑制剂结合,而极性溶解自由能和熵变贡献不利于抑制剂的结合.抑制剂8Q9和8QC与BRD4(1)的范德华相互作用分别为-40.68±0.26,-37.54±0.24 kcal·
mol-1,这为抑制剂与BRD4(1)的结合提供了有利的因素.抑制剂8Q9和8QC与BRD4(1)的静电相互作用分别为-17.34±0.39,-22.32±0.57 kcal·mol-1,8Q9/BRD4(1)和8QC/BRD4(1)的极性溶解自由能分别为35.43±0.42、33.61±0.48 kcal·mol-1,可以看到不利的极性溶解自由能完全屏蔽了有利的静电相互作用,进一步产生了不利的相互作用(△Gele+pol),从而严重削弱了两种抑制剂与BRD4(1)的结合.此外,尽管非极性溶解自由能也为抑制剂结合提供了有益的作用力,但非极性溶解自由能比范德华相互作用弱得多.因此,范德华相互作用在抑制剂与BRD4(1)的结合中起着重要作用.
3.5 抑制剂-残基相互作用
基于残基的自由能分解的分析可以阐明单个残基在抑制剂结合中的作用.为了进一步了解各个分离残基与抑制剂的结合,将两个抑制剂与BRD4(1)复合体的总结合自由能分解为每个残基的贡献(图5),并且采用Amber18中的CPPTRAJ模块分析了抑制剂与BRD4(1)的氢键相互作用(表2).图6和图7采用动力学模拟轨迹生成的最低能级结构,描绘了关键残基相对于抑制剂的几何位置.

表2 动力学模拟过程中抑制剂与关键残基形成的氢键Table 2 The hydrogen bonds formed between key residues and inhibitors.

图5 抑制剂与BRD4(1)中各分离残基的相互作用谱:(a)8Q9/BRD4(1),(b)8QC/BRD4(1)Fig.5 Interactions between two inhibitors and separate residues of BRD4(1):(a)the 8Q9/BRD4(1),(b)the 8QC/BRD4(1).

图6 抑制剂相对于BRD4(1)的关键残基的几何位置:(a)8Q9/BRD4(1),(b)8QC/BRD4(1).Fig.6 Geometric positions of inhibitors relative to key residues of BRD4(1)involving strong interaction:(a)8Q9/BRD4(1),(b)8QC/BRD4(1).

图7 抑制剂与BRD4(1)中关键残基的氢键相互作用:(a)8Q9/BRD4(1),(b)8QC/BRD4(1).Fig.7 Hydrogen bonds formed between two inhibitors and key residues of BRD4(1):(a)8Q9/BRD4(1),(b)8QC/BRD4(1).
图5(a)表明六个残基与8Q9的相互作用强于1 kcal·mol-1,这些残基是Trp81,Pro82,Val87,Leu92,Leu94和Ile146.Ile146是这些残基中和抑制剂相互作用最强的残基,相互作用能是-3.30 kcal·mol-1,主要来自Ile146烷基与抑制剂的疏水环的CH-π相互作用(图6(a)).残基Trp81和Pro82与抑制剂的相互作用能分别是-1.48和-1.34 kcal·mol-1,该相互作用源自残基的疏水环临近抑制剂的疏水环从而产生的π-π相互作用.残基Val87,Leu92和Leu94因其各自的烷基靠近抑制剂的疏水环从而产生CH-π相互作用,分别贡献了-1.70,-2.14和-1.57 kcal·mol-1的作用能.
图5(b)表明六个残基与8QC的相互作用强于1 kcal·mol-1,这些残基是Trp81,Pro82,Lys91,Leu92,Cys136和Ile146,其中残基Ile146和抑制剂相互作用最强,相互作用能是-3.45 kcal·mol-1,该相互作用在结构上吻合8QC的疏水环与Ile146烷基的CH-π相互作用(图6(b)).残基Trp81和Pro82与抑制剂的相互作用能分别是-1.14和-1.59 kcal·mol-1,该相互作用是两个残基的疏水环与抑制剂的疏水环产生的π-π相互作用.残基Leu92的烷基靠近抑制剂的疏水环从而产生CH-π相互作用,且贡献了-2.23 kcal·mol-1的相互作用能.上述分析表明疏水相互作用(例如CH-π和π-π相互作用)在抑制剂与BRD4(1)的结合中起关键作用.
氢键对于抑制剂与蛋白质的结合是必不可少的.在我们的研究中,基于最后80 ns的分子动力学模拟轨迹,采用Amber18中的CPPTRAJ模块,计算了抑制剂与关键残基间的氢键距离,角度和占有率(表2和图7).如表2所示,抑制剂8Q9与残基Asn140形成两个氢键相互作用,其对抑制剂结合的能量贡献为-0.81 kcal·mol-1(图5(a)),两个氢键的占有率分别为95.75和56.03%,根据图7,该氢键作用源自残基Asn140的NH基团与8Q9的疏水性环中的氧原子(OAG)及氮原子(NAH)的相互作用;抑制剂8QC与残基Gln85,Lys91和Cys136形成三个氢键相互作用,其对抑制剂结合的能量贡献分别为-0.76,-1.62和-1.41 kcal·mol-1(图5(b)),三个氢键的占有率分别为41.71,59.73和56.46%,根据图7,三个氢键作用分别源自残基Gln85的NH基团与8QC的氧原子(OAM),残基Lys91的氧原子与8QC的NH基团,以及残基Cys136的SH基团与8QC的氮原子(NAH)的相互作用.
鉴于上述分析,疏水性相互作用(例如π-π相互作用和CH-π相互作用)在抑制剂与BRD4(1)的结合中起关键作用.此外,氢键相互作用也是抑制剂与BRD4(1)结合的重要因素.常见残基Trp81,Pro82,Gln85,Val87,Lys91,Leu92,Leu94,Cys136,Asn140和Ile146利于抑制剂与蛋白质的结合,这些残基在设计针对BRD4(1)的有效抑制剂时需要特别关注.因此,在开发针对BRD4(1)的高效抑制剂时,应特别注意疏水相互作用和氢键相互作用.
4 结 论
本研究对apo/BRD4(1),8Q9/BRD4(1),8QC/BRD4(1)三个系统进行了200 ns的分子动力学模拟,以揭示抑制剂与BRD4(1)的结合方式.RMSFs结果表明抑制剂的结合对BRD4(1)的结构柔性产生了重大影响.动态相关性分析表明抑制剂的结合会导致BRD4(1)的运动模式发生重大变化.借助MM-GBSA方法有效地估算了两种抑制剂对BRD4(1)的结合自由能,计算结果的趋势与实验值吻合良好.同时,结合自由能结果表明范德华相互作用在抑制剂与BRD4(1)的结合中起着重要作用.自由能分解的结果表明残基Trp81,Pro82,Val87,Leu92,Leu94和Ile146主要与抑制剂产生疏水相互作用(例如π-π相互作用和CH-π相互作用),而残基Gln85,Lys91,Cys136和Asn140与抑制剂产生了氢键相互作用,因此,这些残基在今后设计靶向BRD4(1)抑制剂时应特别注意.综上所述,以上结果表明控制抑制剂与BRD4(1)结合的主要是疏水性相互作用和氢键相互作用.我们希望这项研究可以为以后开发抑制BRD4活性的有效抑制剂提供有用的信息.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
科学技术创新(2022年36期)2022-02-13
科学技术创新(2022年36期)2022-02-01
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
中国机械工程(2019年7期)2019-04-23
都市家教·上半月(2017年7期)2017-08-15
中学化学(2015年12期)2016-01-19
池州学院学报(2015年3期)2016-01-05
天津科技大学学报(2015年2期)2015-08-09
原子与分子物理学报(2014年3期)2014-02-28
