
4,7-二(5-溴噻吩-2-基)-2,1,3-苯并噻二唑低聚物光伏性能的理论研究
2021-08-16 07:26张福兰
原子与分子物理学报 2021年3期
张福兰
(长江师范学院化学化工学院重庆市无机特种功能材料重点实验室,重庆 408100)
1 引 言
随着能源的不断耗竭,寻找清洁、可再生能源已迫在眉睫.太阳能,取之不尽用之不竭,照射到地球上1小时的能量大约相当于全球1年的能耗[1],因此,有效利用太阳能对人类可持续发展具有重要的意义.太阳能电池是有效利用太阳能的途径之一,自1954年贝尔实验室研制出第一个硅太阳能电池以来,经过数十年的研究发展,太阳能电池已经历了三代[2].近十年来,作为一种潜在可再生能源的有机聚合物太阳能电池已被人们广泛使用,因为有机聚合物太阳能电池具有材料易得、材质轻、制备工艺简单而耗资低、可大面积制成柔性膜等优点而成为人们关注的热点[3,4].有机聚合物构型是影响有机聚合物太阳能电池转换效率的关键因素之一,目前有机聚合物单体的研究报道也非常多[5-8].4,7-二(2-噻吩基)苯并噻二唑(DTBT)是目前最受关注的有机聚合物单体,已有很多从实验、分子掺杂等方面的研究报道[9-13].Wang等[14]从分子间作业角度讨论了F代DTBT的光伏性能.Chen等[15]从理论角度研究过杂原子在DTBT分子内桥接后的光伏性能.也有对于低聚合物相关性质的研究报道:夏琼等[16]从理论角度讨论过PC-DTBT低聚合度时的相关性质.Benatto等[17]从理论角度研究了DTBT苯环H被F原子取代后对有机分子受体的激子结合能和电荷传输的影响性能.目前,虽然有诸多关于DTBT多个性能的相关报道,但对于溴代DTBT低聚合度相关性质的理论研究还未见报道,因此,本文采用密度泛函理论(DFT)方法研究了DTBT的噻吩环上一个H原子被卤素Br原子取代以后的化合物(简称DT(Br)BT,结构如图1所示)在低聚合度(DT(Br)BT)n(n=1~6)的光伏性能,取得了很好的研究结果,希望我们的研究能为探究有机聚合物在太阳能电池材料方面的应用提供一定的帮助.
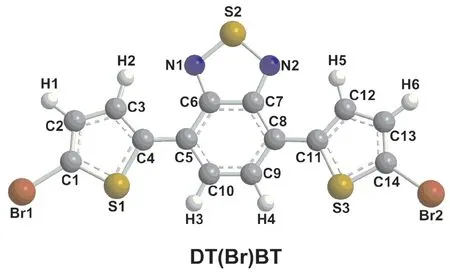
图1 DT(Br)BT几何优化结构Fig.1 Optimized geometric structure of DT(Br)BT
2 计算方法
采用DFT研究了(DT(Br)BT)n(n=1~6)的稳定构型、前线轨道和吸收光谱.计算过程中采用广义梯度近似(GGA)中Perdew-Wang-91(PW91)[18]函数中DNP[19]基组,原子序数小于21的原子的全部电子参与计算,即采用全电子基组(All electron),S和Br原子采用effective core potentials(ECPs)[19]赝势,Quality设为fine,拖尾效应值选为0.005 Ha,优化收敛精度取程序内定值.全部计算工作在Materimals Studio 5.5软件中以DFT为基础的Dmol3程序包[20,21]进行.
3 结果和讨论
3.1 构型稳定性分析
优化得到DT(Br)BT的键长参数如表1所示.由表1中的数据可以看出,化合物DT(Br)BT中的C-C单键和C=C双键键长在0.1370~0.1457 nm之间,C-N键长为0.1347 nm,C-Br键长为0.1899~0.1905 nm之间,C-S键长为0.1730~0.1765 nm之间,N-S键长为0.1641~0.1642 nm之间,C-H键长为0.1085~0.1090 nm之间,这些键长都在正常键长范围,说明我们选择的计算方法可靠.
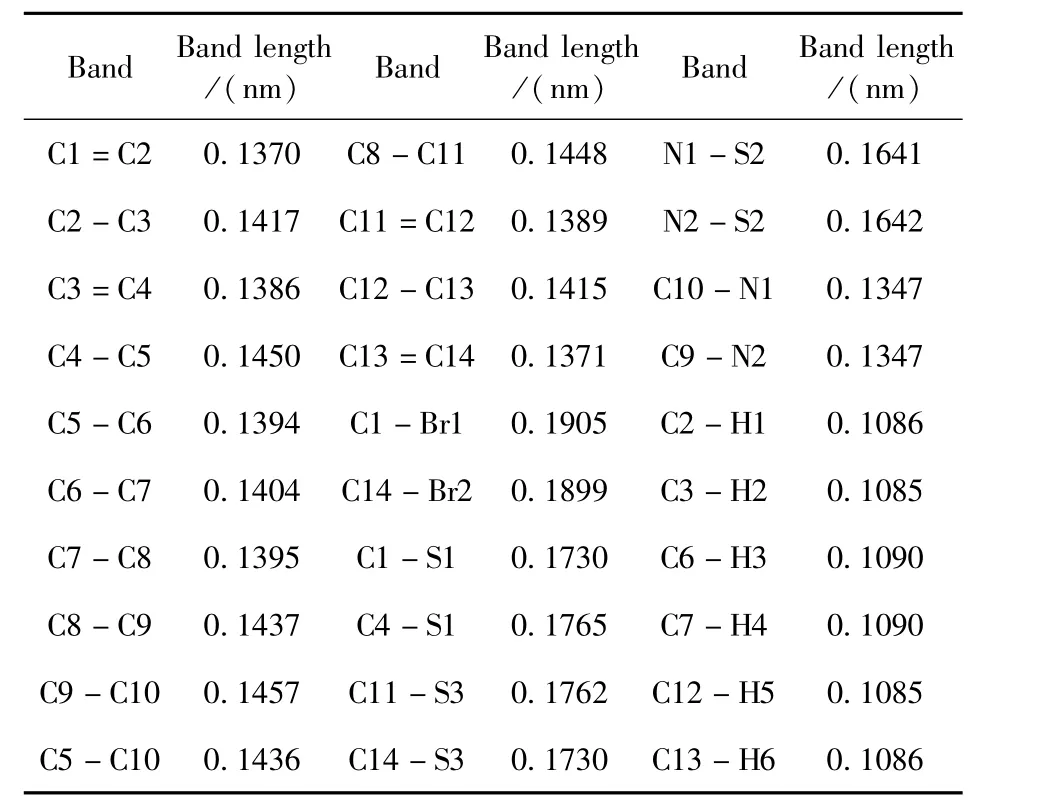
表1 DT(Br)BT中各键键长Table 1 Various band lengths(nm)in steady structure of DT(Br)BT
图2给出了(DT(Br)BT)n(n=1~6)分子构型的侧视图和俯视图.图2(a)为DT(Br)BT单体结构,从侧视图可以看出噻吩环与苯并噻二唑环不在一个平面内,旋转的角度分别为2.9˚(2.03˚[11])和3.1˚(4.26˚[11]),数据显示与已有的报道结果一致.从图2(b)可以看出,DT(Br)BT的二聚体所有的环几乎在同一平面且形成一个大π键,这很有利于电子传输.当3个DT(Br)BT分子聚合时,第3个分子与另外两个分子构成的平面成32.5˚的二面角,而当4个DT(Br)BT分子聚合时,首尾分子与中间两个分子构成的平面分别成35.8˚和16.2˚的二面角,形如小船(图2(d)).从图2(e)可以看出,当5个DT(Br)BT分子以链状聚合时,中间有两个分子在同一平面,一端一个分子与中间平面成9.5˚的二面角,而另一端有两分子形成一个平面,且这个平面与中间平面成43.3˚的二面角.而当6个DT(Br)BT分子以链状聚合时,整个大分子呈“S”型,首尾分子与中间两个分子构成的平面分别成32.8˚和16.6˚的二面角(图2(f)).由于环状分子比链状分子稳定,图2(g)和(h)分别是DT(Br)BT的环状五聚体和环状六聚体,可以看出,DT(Br)BT的环状五聚体为平面构型,而环状六聚体的构型类似于苯环的船型结构,两船头上翘的二面角分别为12.4˚和10.3˚.由于有机聚合物分子结构的平整性是直接影响太阳能电池材料填充因子的关键因素之一[22],通过对比(DT(Br)BT)n(n=1~6)的构型可以发现,链状二聚体和环状五聚体为平面骨架,是比较理想的光伏材料前驱体.
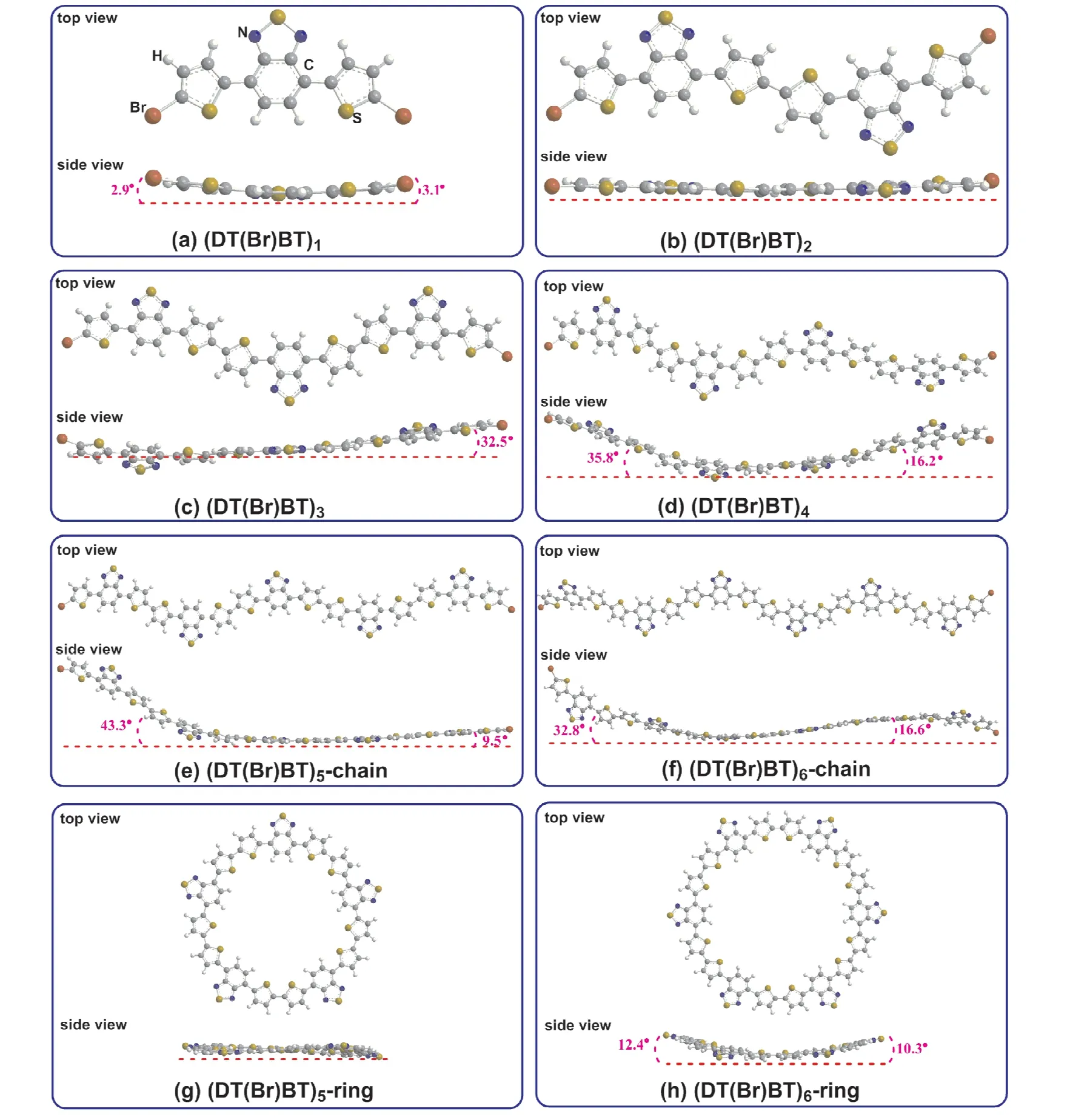
图2 (DT(Br)BT)n(n=1~6)的优化构型示意图.Fig.2 Optimized geometric structures of(DT(Br)BT)n(n=1~6).
3.2 前线轨道分析
图3 给出了(DT(Br)BT)n(n=1~6)的最高占据轨道(HOMO)和最低未占据轨道(LUMO)的电子云示意图.由图3(a)可以看出,DT(Br)BT的HOMO电子云聚集在碳碳骨架结构的离域π键上,LUMO电子云主要聚集苯并噻二唑上,这和之前报道的文献研究结果非常一致[10,11,13,17,23,24].由图3(b)可以看出,DT(Br)BT二聚体的HOMO电子云聚集位置与单体一样,只是电子云分布链更长,而LUMO电子云除了聚集在两个噻二唑环上以外,还在两个苯环和中间两个噻吩环之间形成了一个长链电子云,这主要是由于二聚体分子中有为平面构型的离域大π键的缘故.由图3(c~f)可以看出,(DT(Br)BT)n(n=3~6)的链状聚合物的前线轨道电子云聚集位置主要在分子骨架中间部分,而且随着聚合物的聚合度增大,电子云向中间聚集的程度越大,首尾变得越来越弱.再对比图3(g)和(h)可以看出,环状五聚体的前线轨道电子云完全连续均匀分布在大环分子骨架上,而环状六聚体的前线轨道电子云在大环分子骨架上出现了不连续性,这可能是由于(DT(Br)BT)5-ring为平面构型而(DT(Br)BT)6-ring为船型的缘故.由此可见,链状二聚体和环状五聚体具有相对较好的光伏性能,与前面研究结论一致.
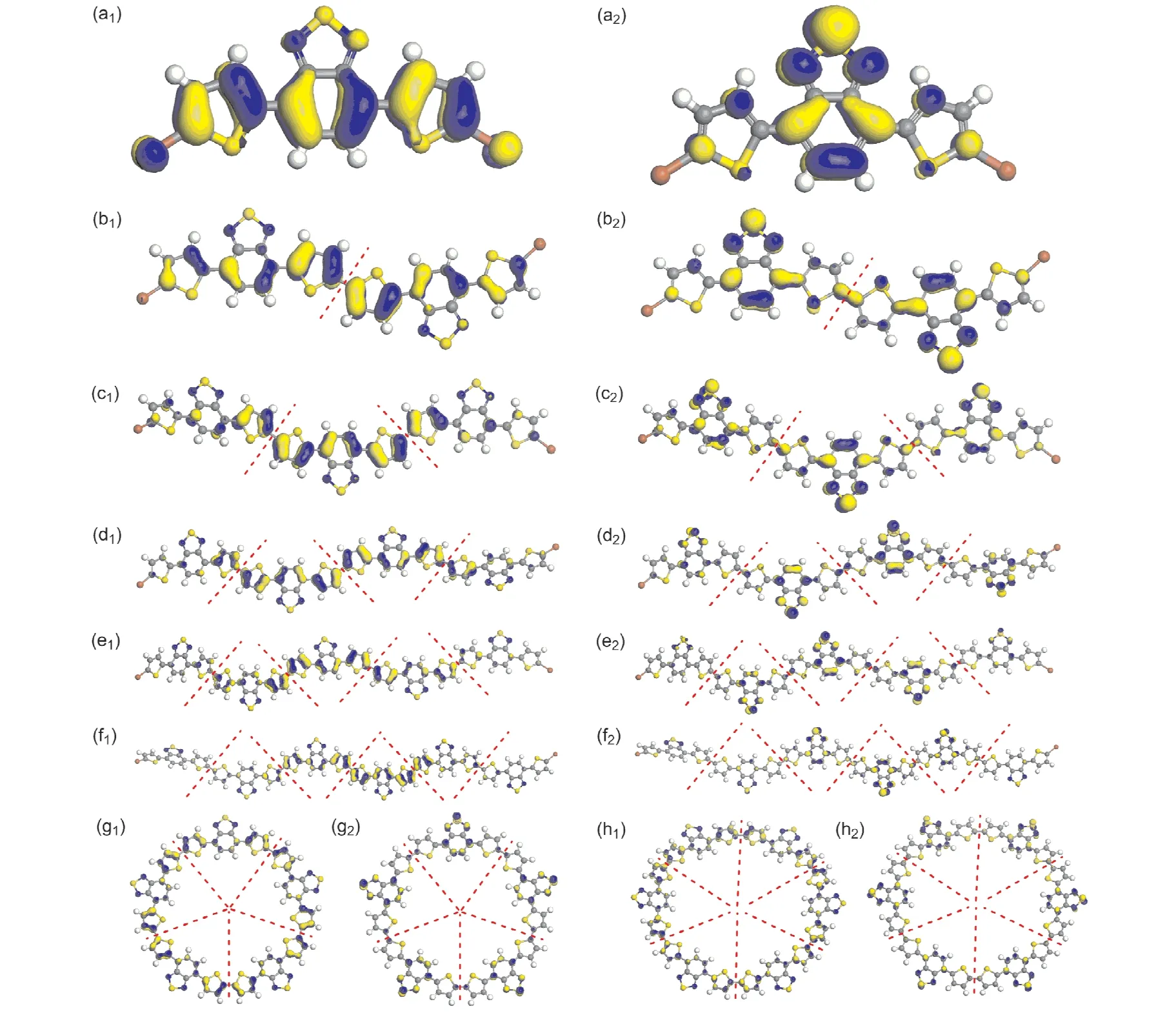
图3 最高占据轨道(HOMO)和最低空轨道(LUMO)的电子云示意图.(下标为1的是HOMO,下标为2的是LUMO)(a)(DT(Br)BT)1,(b)(DT(Br)BT)2,(c)(DT(Br)BT)3,(d)(DT(Br)BT)4,(e)(DT(Br)BT)5-chain,(f)(DT(Br)BT)6-chain,(g)(DT(Br)BT)5-ring,(h)(DT(Br)BT)6-ring.Fig.3 Electron density plots of the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO).(The subscripts“1”represent HOMO,the subscripts“2”represent LUMO.)(a)(DT(Br)BT)1,(b)(DT(Br)BT)2,(c)(DT(Br)BT)3,(d)(DT(Br)BT)4,(e)(DT(Br)BT)5-chain,(f)(DT(Br)BT)6-chain,(g)(DT(Br)BT)5-ring,(h)(DT(Br)BT)6-ring.
表2列出了(DT(Br)BT)n(n=1~6)的总能量、聚合能、前线轨道能和能隙(Eg).链状分子的聚合能通过下面方程(1)计算得到,环状分子的聚合能通过下面方程(2)计算得到[16]:
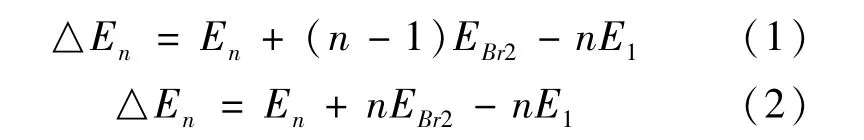
其中,△En为(DT(Br)BT)n的聚合能,En代表聚合物(DT(Br)BT)n的能量,E1代表(DT(Br)BT)1的能量,EBr2代表小分子Br2的能量,n(n=1~6)为聚合度.
由表2可以知道,(DT(Br)BT)n(n=2~6)的Eg均比单体DT(Br)BT的Eg小,并且随着n逐渐变大,Eg逐渐降低,当n≥3时,Eg<1.00 eV,变化趋势更小.从表2可知,形成(DT(Br)BT)n的△En均大于0,同时(DT(Br)BT)n的△En随着n的增加而逐渐增大,表明它们的稳定性减弱.研究发现当(DT(Br)BT)2的Eg为1.11 eV,与硅的带隙相当(硅的带隙大约在1.1 eV[2]),这点也说明(DT(Br)BT)2有很好的光伏活性.

表2 总能量(E total/a.u.)、聚合能(△E/eV)、前线轨道能和能隙(E g/eV)Table 2 Energies(E total/a.u.),polymer energies(△E/eV),energies of HOMO(E HOMO/eV)and LUMO(E LUMO/eV),and energy gaps(E g/eV)of all the configurations.
3.3 吸收光谱分析
为了进一步阐明(DT(Br)BT)n的光伏性能,用DFT模拟了(DT(Br)BT)n的吸收光谱.如图3(a),(DT(Br)BT)1有两个不同的吸收带,最强的一个吸收峰在高能紫外区域(350~400 nm)的384 nm处,这源于局部的π-π*跃迁所致[24],而另一个弱吸收峰在低能可见光区域(530~580 nm)的574 nm处,这是典型的分子内电荷迁移所引起的跃迁[24].如图3(b),(DT(Br)BT)2有两个不同的吸收带,高能区域吸收带(430~580 nm)分别在463,505和553 nm处出现三个吸收峰,最强吸收峰在低能区域吸收带(730~770 nm)的757 nm处.如图3(c~f),(DT(Br)BT)n(n=3~6)链状聚合物的最强吸收峰分别为909,1011,1111和1152 nm处,都在红外区.对比图3(g)和(h)可知,(DT(Br)BT)5-ring在红外区844 nm处有唯一很强的吸收峰,这是由于(DT(Br)BT)5-ring分子内形成了首尾相连的共轭大环平面,只存在分子内电荷迁移跃迁,这类似于本研究小组曾报道过用3个Si原子掺杂DTBT衍生物A4B7BT中苯环C间位所的化合物在可见光区有唯一强吸收峰一样[25];而(DT(Br)BT)6-ring在红外区有两个吸收峰,最强在937 nm处.经过数据分析还可看出,DT(Br)BT单体的最强吸收峰在紫外区,而(DT(Br)BT)n(n=2~6)最强吸收峰均发生红移,只有(DT(Br)BT)2最强吸收峰在可见光区(400~760 nm[26]),其余(DT(Br)BT)n(n=3~6)的最强吸收峰却都在红外区,而且环状聚合物红移程度不如链状聚合物红移程度大.综合前述构型、前线轨道和吸收光谱可知,在可见光区,DT(Br)BT的二聚体光伏性能最好;而在红外区,(DT(Br)BT)5-ring的光伏性最好.

图3 (DT(Br)BT)n(n=1-6)的吸收光谱示意图.(a)(DT(Br)BT)1,(b)(DT(Br)BT)2,(c)(DT(Br)BT)3,(d)(DT(Br)BT)4,(e)(DT(Br)BT)5-chain,(f)(DT(Br)BT)6-chain,(g)(DT(Br)BT)5-ring,(h)(DT(Br)BT)6-ring.Fig.3 Absorption spectra of the(DT(Br)BT)n(n=1-6).(a)(DT(Br)BT)1,(b)(DT(Br)BT)2,(c)(DT(Br)BT)3,(d)(DT(Br)BT)4,(e)(DT(Br)BT)5-chain,(f)(DT(Br)BT)6-chain,(g)(DT(Br)BT)5-ring,(h)(DT(Br)BT)6-ring.
4 结 论
采用DFT方法,在PW91/DNP水平上研究了低聚合物(DT(Br)BT)n(n=1~6)的稳定构型、前线轨道和吸收光谱.稳定构型和前线轨道分析表明,随着聚合度n增加,(DT(Br)BT)n的聚合能增强,前线轨道能隙变窄,而且DT(Br)BT的链状二聚体和环状五聚体均为平面构型.光谱分析表明,DT(Br)BT单体的最强吸收峰384 nm在紫外区,而(DT(Br)BT)n(n=2~6)最强吸收峰均发生红移,且环状聚合物红移程度不如链状聚合物红移程度大,(DT(Br)BT)2最强吸收峰757 nm在可见光区,其余(DT(Br)BT)n(n=3~6)的最强吸收峰却都在红外区.由此可见,DT(Br)BT的链状二聚体和环状五聚体具有很好的光伏性能,都是很有希望的有机太阳能电池材料前驱体.
猜你喜欢
化学教学(2022年4期)2022-05-07
读写算(2020年32期)2020-12-17
中南民族大学学报(自然科学版)(2020年2期)2020-04-18
丝路视野(2018年1期)2018-05-14
中学课程辅导·教师教育(上、下)(2017年12期)2017-07-01
临床医药文献杂志(电子版)(2017年95期)2017-03-20
中国卫生标准管理(2015年1期)2016-01-14
五邑大学学报(自然科学版)(2015年3期)2015-10-20
河北科技大学学报(2015年6期)2015-03-11
河北科技大学学报(2015年5期)2015-03-11
