
噻吩并嘧啶衍生物抗胃癌活性的CoMFA模型与分子设计
2021-08-16 07:26唐自强冯长君
原子与分子物理学报 2021年3期
唐自强,冯 惠,冯长君
(1.徐州工程学院材料与化学工程学院,徐州 221018;2.徐州技师学院,徐州 221151)
1 引 言
噻吩并嘧啶衍生物是一类具有杀菌、抗过敏、除草、抗惊厥和抗癌等良好生物活性的稠杂环化合物,具有较好的临床开发价值,特别是其抗癌活性引起了人们的重视并展开了相关的研究[1].王红梅等[2]应用5,6,7,8-四氢苯并噻吩膦亚胺,经氮杂Wittig反应合成了不同取代的新型稠合噻吩并[2,3-d]嘧啶-4(3H)-酮衍生物,通过体外抗肿瘤活性测试,显示这些化合物具有良好的抗肿瘤活性.周新等[3]以1,2,4-三氮唑为起始原料,经Gewald反应、Wittig反应合成了含氟噻吩并嘧啶酮类衍生物,其抑菌活性测试结果表明它们对棉花枯萎菌、水稻纹枯菌、黄瓜灰霉菌、小麦赤霉菌、苹果轮纹及棉花炭疽具有较好的抑制作用.近年来研究发现,吡啶并嘧啶衍生物如吡啶并[4,3-d]嘧啶化合物也是一类具有良好抗肿瘤活性的杂环化合物,并对多种肿瘤细胞均有较强的抑制作用.这些噻吩并嘧啶、吡啶并嘧啶类杂环化合物不仅能够抑制多种肿瘤细胞的形成、增殖,而且还可以诱导细胞的凋亡,有望成为新型的抗肿瘤药物.
基于噻吩并嘧啶、吡啶并嘧啶骨架展现出的优异抗肿瘤活性,王胜红等[4]应用氮杂Wittig反应合成了25个嘧啶环上具有不同取代基的新型四氢苯并[4',5']噻吩并3',2':5,6]吡啶并[4,3-d]嘧啶衍生物,简称“噻吩并嘧啶衍生物”.他们以5-氟尿嘧啶(5-FU)为阳性对照药,采用MTT法对合成的化合物进行抗胃癌细胞(MGC-803)活性评价.
物质构效关系(quantitative structure-activity relationship,QSAR)[5-11]是研究化合物分子结构与生物活性之间因果关系,揭示其间量变规律,并利用规律估算与预测活性,以及探讨作用机理等.其在预测化合物生物活性方面已成为化学、医药、环境等学科的一个重要领域.本文采用比较分子力场分析(comparative molecular field analysis,CoMFA)方法[12-19]研究噻吩并嘧啶衍生物对胃癌的抑制活性(p M)[4],以揭示影响它们抗肿瘤活性的微观结构因素,探讨其抑制肿瘤作用的分子机理.
2 实验方法
2.1 数据来源
王胜红等[4]合成的25种噻吩并嘧啶衍生物的分子基本结构见图1.其中取代基R1、R2为分子中可变部分,具体分子结构见表1.他们测定抗胃癌细胞的半数抑制浓度以“IC50”表示,单位为μmol·L-1.考虑化学反应自由能变与平衡浓度为对数关系,故令:
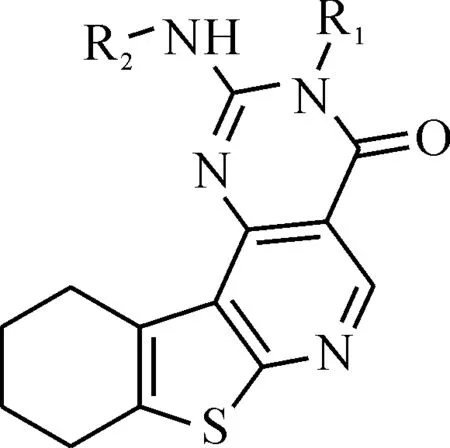
图1 噻吩并嘧啶衍生物分子的基本结构Fig.1 Basic structure of thienopyrimidine derivative molecules

它们的IC50及p M数据见表1.
2.2 建模方法
本文3D-QSAR[17]分析及建立CoMFA模型均使用Tripos公司Sybylx2.1.1分子模拟软件完成,使用的模块包括SYBYL、Sketch、Minimize、Database-alignment以及CoMFA的QSAR方法等,各项参数除特别指明外均采用缺省值.
2.2.1 化合物低能构象的确定及分子叠合
分子活性构象的确定是建立有效3D-QSAR模型的前提之一.通常采用分子的最低能量构象代替药效活性构象.使用Sybylx2.1.1软件中的Sketch molecule模块先构建25个噻吩并嘧啶衍生物分子的初始三维结构,再通过Minimize模块,选取Tripos力场,加Gasteiger-Huckel电荷,将最大迭代次数(Max.Iterations)定为1000,将Gradient降低到0.005;最后将Powell能量梯度法的收敛梯度设为0.21kJ·mol-1·nm-1.对所有分子进行分子力学能量最低优化,通过全构象搜索得到能量最低构象,并以此最低能量构象作为分子叠合的生物活性构象.
随机选取分子3、9、14、17、21为测试集(Test set;含13号分子.为表1中带“*”的分子),余下20个分子作为训练集(Training set).选取抗胃癌活性最强的13号为模板分子,采用公共骨架的叠合方式(即保证所有分子取向的一致性,使分子间相互重叠时的均方根偏差最小)予以叠合.即以13号分子中的公共骨架,运用Align database模块分别对训练集、测试集进行叠合.训练集的叠合图见图2(注:测试集的叠合图与其非常相似不予列出):
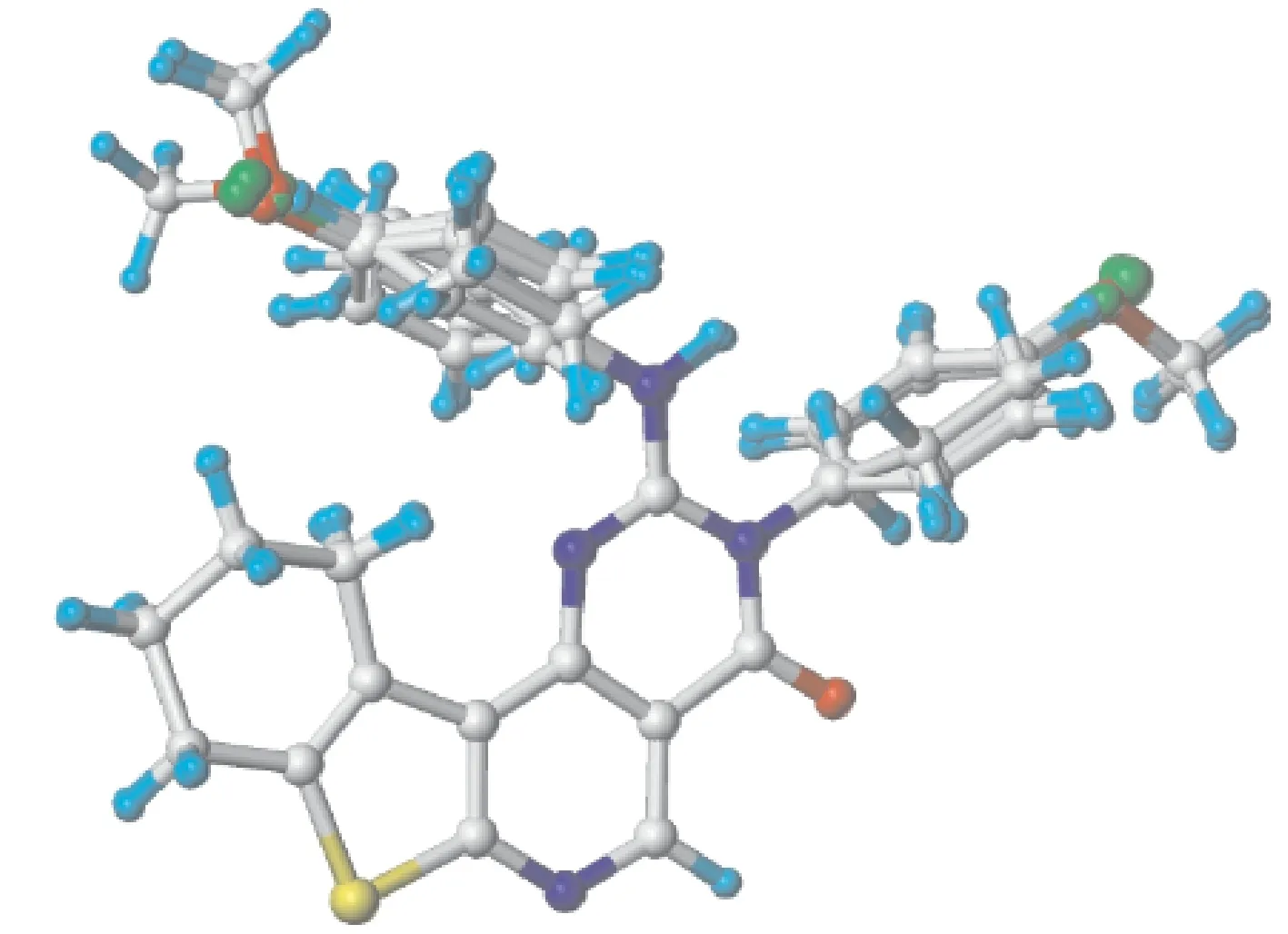
图2 训练集的叠合图Fig.2 3D view of all the aligned molecules in training set

表1 噻吩并嘧啶衍生物分子结构与抗胃癌活性(p M)Table 1 The molecular structures and anti-gastric cancer activity(p M)of thienopyrimidine derivatives
2.2.2 CoMFA模型的建立
采用Tripos标准力场,对训练集的叠合分子周围每个网格点上的立体场(Steric,St)及静电场(Electrostatic,El)予以计算,以偏最小二乘法(Partial least squares,PLS)进行建模.采用逐一剔除法(Leave-one-out,LOO)进行交叉验证,以获交叉验证系数Rcv2和最佳主成分数N.一是以Rcv2衡量模型的预测能力,要求Rcv2>0.3的模型,具有95%的可信度[20],随机性小于5%.二是模型中的化合物数(m)与变量数(N)之比,即样变比S v,S v≥5才具有统计意义,和良好的稳健性及较低或然性[21].三是进行回归分析,得到非交叉验证判定系数R2、统计方差比F.最后采用View CoMFA模块,以三维等势图直观反映立体场和静电场对噻吩并嘧啶衍生物p M的贡献.
3 结果与讨论
3.1 CoM FA模型
训练集的CoMFA模型:(1)交叉验证部分:Rcv2=0.397>0.3,N=4,S v=m/N=5,表明模型具有很好的预测能力与稳健性.(2)非交叉验证部分:R2=0.835>0.8,显示良好拟合性;R2又称为削减误差比例,意为该模型包含影响噻吩并嘧啶衍生物抗胃癌活性83.5%的因素,仅有不足16.5%的属于未知因素.(3)在95%显著水平下,F临界值为:F0.05(4,14)=3.11.该模型的F=18.95,是其临界值的6倍多,表明模型是密切相关,具有统计学稳定性.因此,该模型具有较低的标准偏差SE,仅为0.333.利用训练集的3D-QSAR模型对测试集中分子的p M进行预测,以检验其预测能力.测试集的p M预测值见表1,与相应实验值基本吻合,其平均误差只有0.180,表明模型确实具有良好的预测能力.25个化合物的实验值与估算值(及预测值)的散点图见图3,这些散点基本都密集在直线附近,证明该模型具有良好的相关性及预测能力.
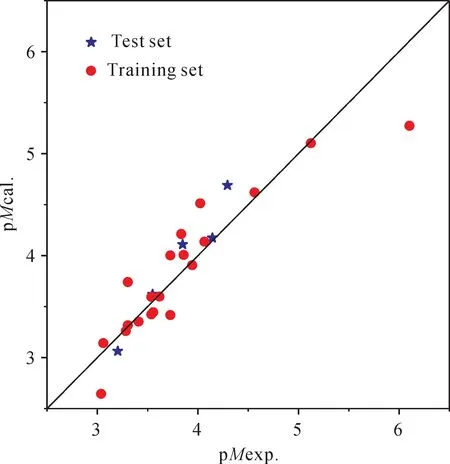
图3 噻吩并嘧啶衍生物抗胃癌活性的实验值与预测值的相关图Fig.3 Plot of the experimental p M vs.calculated p M of thienopyrimidine derivatives
3.2 CoM FA等势图
图4 给出了以训练集中抗胃癌活性最高的13号分子为模板建立的CoMFA模型的三维等势图,分子周围不同颜色的块状图表示噻吩并嘧啶衍生物分子中取代基的立体场和静电场对抗胃癌活性的影响.图4(a)为立体作用等势图,增大取代基体积有利于提高噻吩并嘧啶衍生物抗胃癌活性的区域以绿色表示,黄色区域表示减小取代基体积对抗胃癌活性有利.由立体场的空间分布可知,在2个苯环的3、4-位上(较大绿色区域)引入较大体积的基团,有助于提高噻吩并嘧啶衍生物抗胃癌活性.如在R1相同下,4-MeO-Ph、-Ph、n-C3H7的体积依次减小,它们的疏水性降低,透过胃癌细胞膜难度增大,进入细胞内浓度减小,与胃癌靶标作用几率下降,导致p M(实验值)亦是逐渐降低.
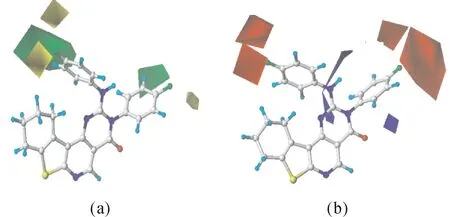
图4 CoMFA模型的立体场(a)与静电场(b)等势图Fig.4 CoMFA contour maps(a)steric field;(b)electrostatic field
图4 (b)为化合物周围的静电场分布,增大基团正电性有利于抗胃癌活性增强的区域以蓝色表示,红色区域则表示引入带负电荷的基团对活性有利.由静电场的空间分布可知,仅有远离苯环的2、3位之间小块蓝色区域,可以忽略其对抗胃癌活性的影响.而在2个苯环的4-位上都被大片红色区域覆盖,说明在这些区域引入负电性基团,有助于提高噻吩并嘧啶衍生物的抗胃癌活性.因此,在R1相同下,4-F、4-MeO、4-Cl的吸电子能力递降,它们的p M亦是逐减.例如13、12、14的p M为6.105、5.125、4.295,依次减小.
训练集的3D-QSAR模型给出的立体场和静电场对p M的贡献分别为40.9%和59.1%,显示取代基的立体作用弱于静电作用.即在R1(或者R2)相同下,p M的主要取决于R2(或者R1)中苯环上取代基的吸电子能力.例如4-MeO、4-Cl、4-F的体积是依次减小,而吸电子能力递增顺序为4-Cl、4-MeO、4-F,因此,它们的p M按此顺序渐增.可见,4-F是最大的负电性基团,故其抗胃癌活性最强,此与文献[4]结果一致.由于静电作用一般包含库仑力、氢键及配位,立体作用对应疏水性和空间位阻.因此,抗胃癌活性主要源于取代基与胃癌细胞内生物受体之间的库仑力、氢键及配位,其次是疏水性和空间位阻.尚需指出,13号分子的R1、R2均为4-F,其p M高达6.105.比第二强的12号分子(其R1为4-F,R2为4-MeO)高出0.980.而CoMFA模型给出13、12号分子预测值为5.270、5.099,相差仅为0.171.有理由认为此0.980中可能包含均为4-F的强强联合的附加效应,也可能存在较大的实验误差.
3.3 分子设计
QSAR研究目的之一是根据所建模型中隐含的影响生物活性的主要结构信息进行分子设计.由3.2分析可知,在R1、R2中苯环的4-位上引入体积较大的负电性基团,有利于增强噻吩并嘧啶衍生物的抗胃癌活性.据此设计4个化合物(见表1中第26-29号化合物),3D-QSAR模型给出较大的p M预测值,在5.430-5.844之间,均优于13号分子预测值(5.270).这些理论上优良的抗胃癌化合物,有待生物医学实验确认.
4 结 论
(1)采用比较分子力场方法(CoMFA)建立噻吩并嘧啶衍生物的抗胃癌活性的3D-QSAR模型:Rcv2=0.397,S v=5,R2=0.835,F=18.95,显示良好的稳定性、相关性和预测性.
(2)CoMFA模型三维等势图显示在R1、R2中苯环的4-位上引入体积较大的负电性基团,有利于增强噻吩并嘧啶衍生物的抗胃癌活性.据此设计的4个化合物,具有较大的p M预测值,有待生物医学实验确认.
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
武警医学(2018年10期)2018-11-06
温州大学学报(自然科学版)(2016年1期)2016-10-27
当代化工研究(2016年6期)2016-03-20
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
应用化工(2014年9期)2014-08-10
应用化工(2014年7期)2014-08-09
无机化学学报(2014年5期)2014-02-28
无机化学学报(2014年3期)2014-02-28
