
植物基因组组装技术研究进展
2021-08-11 02:45唐蝶周倩
生物技术通报 2021年6期
唐蝶 周倩
(1. 中国农业科学院深圳农业基因组研究所,深圳 518124;2. 鹏城实验室,深圳 518055)
基因组组装是将全基因组测序的小片段(read,长度100 bp-100 kb)通过算法拼接成尽量长的片段(contig和scaffold,长度几十kb到Mb不等)或者整条染色体的过程。组装过程的关键是识别基因组上相邻测序片段的重叠关系,除测序技术外,基因组的杂合度和重复序列对组装效果影响最大[1]。植物基因组往往经历局部复制、全基因组加倍、重复序列扩张等,导致基因组中存在大量相似或者同源的片段,组装时产生冗余的重叠关系,增加组装的困难。由于植物基因组具有非常丰富的多样性,参考已发表的少数物种组装新的物种,有时却无法达到理想的组装效果。测序技术发展提供了短序列测序、单分子测序、光学图谱、Hi-C图谱等多种测序技术及其组合的组装方案[2],如何以最低成本获得满足研究需求的基因组,是科研人员普遍面临的一个问题。本文综合阐述植物基因组特征与组装效果之间的关系,以期对研究人员选择组装策略、预估组装结果提供一定的参考。
1 基因组特征评估
在组装前通常需要对基因组进行评估,获得基因组的概括性特征。在各项特征中,基因组大小、杂合度和重复序列含量是决定测序成本、组装难度和最终组装效果的最重要的几个特征[3]。这些特征可以通过全基因组的K-mer分析进行评估。在测序read上相隔1 bp取长度为K的子序列,称为K-mer,全部测序read中K-mer的种类及其出现次数(K-mer深度)通过分布曲线展示出来,即可观察到基因组的基本特征(图1)。在测序覆盖均匀、没有测序错误和重复序列的基因组上,K-mer分布曲线符合泊松分布。如果基因组存在某些复杂特征,会使分布曲线偏离泊松分布,出现与特征相对应的峰。
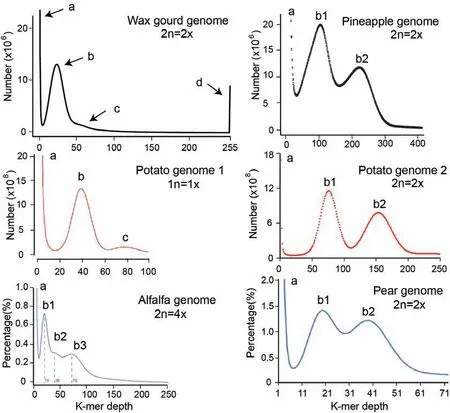
图1 几种植物基因组Illumina测序数据K-mer分布曲线Fig. 1 K-mer volume histograms of illumina sequencing data from several plant genomes
在实际测序数据的K-mer分布曲线上,第一个极高的值是测序错误导致的K-mer,深度只有1-2。单倍体或纯合基因组的K-mer分布曲线只有一个主峰。杂合二倍体基因组的K-mer分布曲线有两个峰,分别为杂合峰和纯合峰,前者深度只有后者的一半。杂合多倍体基因组则会出现多个杂合峰。杂合峰的比例越高,表示杂合度越大。重复序列含量较高时会在主峰后面形成一个小峰或者在极高深度处形成拖尾。
基因组大小可以由(总K-mer数量)/(K-mer期望测序深度)来估计,通常以K-mer分布曲线的主峰深度作为期望测序深度。该公式估算的基因组大小有10%左右的误差,可以结合流式细胞实验检测DNA含量,估算基因组大小进行综合考虑。
2 简单植物基因组组装
基因组大小不超过1Gb,纯合或者杂合度低于千分之五,重复序列含量低于50%的基因组可以被归类为简单基因组。作为模式物种首先完成基因组图谱的拟南芥(Arabidopsis thaliana)、水稻(Oryza sativa L. ssp. indica)等都属于简单基因组。简单基因组使用二代测序数据、二三代测序数据混合或者纯三代测序数据,都可以完成组装(表1)。在二代数据为主的项目中,通常用小片段文库组装contig,大片段文库(mate-pair)构建scaffold;加入少量三代数据混合组装,以填补scaffold中的“空洞”区域。与前两种方式相比,使用纯三代数据组装,能够显著提高组装的连续性、完整性等指标,缩短组装时间。10年前由多国实验室合作、耗费数年完成的马铃薯(Solanum tuberosum L.)[6]和番茄(Solanum lycopersicum)[10]参考基因组,如今由单个团队使用纯三代测序数据就组装了contig N50 提高500倍(32 kb vs 17.3 Mb)[11]和60倍(87 kb vs 5.5 Mb)[12]的新版本。使用三代测序数据获得高质量的组装片段,再利用遗传图谱、Hi-C图谱[13-14]、光学图谱[15]等构建成染色体,是当前解析简单基因组最高效的方案,也是学术期刊对简单基因组组装的普遍要求。

表1 几种植物基因组组装方案及组装结果Table1 Assembly strategies and results of several plant genomes
由于三代测序数据单碱基错误率高达10%-15%,组装得到的基因组通常需要先进行序列纠错(“抛光”)再进行基因注释等分析。基因组纠错可以使用二代数据或者三代数据,必要时两种数据结合进行多次纠错。
3 高杂合基因组组装
自交不亲和和无性繁殖在自然界的植物中普遍存在,造成了基因组的杂合特征。本文讨论的高杂合基因组杂合度约为1%-2%,即同源片段的序列差异达到1%-2%,导致组装时同源区域的read无法充分合并,产生大量分支结构,严重影响组装的连续性及后续分析。
将基因组DNA分成小份分别进行测序、组装是避免杂合片段干扰的一种有效方法[37-38],每份DNA含有极少量杂合片段,基本可作为纯合基因组组装,从而降低组装难度。早期解决杂合基因组使用BAC-by-BAC策略[9],构建数万个BAC克隆,每个单独测序、组装,然后合并成一套基因组。另一种方法是借助减数分裂分离出单套基因组,比如通过花粉培养获得单倍体个体。而对于无法获得单倍体的物种,研究人员则设法从二倍体的测序数据中提取单倍体数据。在杂合菠萝(Ananas comosus(L.)Merr.)基因组项目中,研究人员将杂合菠萝F153与CB5杂交,通过比较后代F1个体与亲本F153的测序read,分离出F153其中一套基因组的read进行组装[5]。
近年来发展的10×Genomeics技术,将大片段DNA分子包裹进油滴添加标签后测序,产生的linked-read保留了基因组长距离的信息,有助于构建更长的scaffold[39]。高杂合杨桃基因组的组装结果显示,单个10×G文库组装的scaffold N50达到2.7 Mb[40],组装指标优于早期杂合梨(Pyrus bretschneideri Rehd.)和菠萝基因组。该方案能以最少的测序和计算成本提供可用的参考基因组,已经在植物基因组中广泛应用。
在早期的基因组项目中,组装的目的是得到一个完整的单倍体参考基因组,因此只取单套基因组进行组装或者将基因组内杂合区域尽量合并。随着对基因组研究的深入,基因组单体型信息越来越受到重视,对杂合物种的基因组提出了分型组装的需求。
Falcon-unzip是最早利用三代测序数据进行杂合基因组组装和分型的工具[35],其组装结果包含一个单倍体参考基因组和杂合区域的局部单体型信息,是目前杂合基因组分型最常见的呈现方式。由于三代测序数据的读长优势,Falcon-unzip组装的杂合物种参考基因组在contig连续性上有显著提升,但是输出的参考基因组混合了两个单体型的序列,在基因注释等后续分析中仍然存在问题。
由于组装算法的局限或变异位点分布不均匀,单纯使用全基因组测序组装的单体型都是局部的、片段化的。借助遗传信息分离同源区域的基因组数据, 再将每个区域组装成单体型,是目前解决高杂合物种组装最成功的方法(图2)。
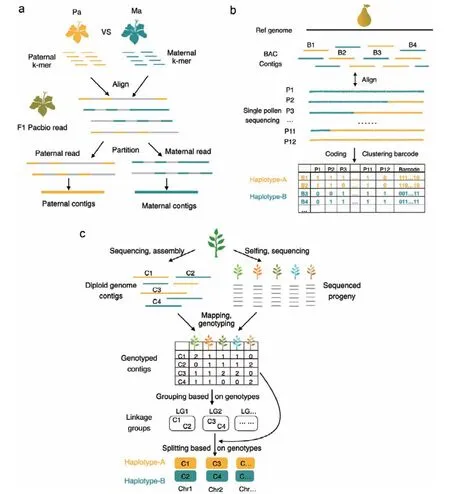
图2 三种植物基因组组装和分型方案Fig. 2 Three assembly and genotyping strategies of genome in plants
“亲本-子代”家系测序是区分杂合个体内两套单体型最直接的方法。Triobin方法将家系测序与第三代测序技术结合[41],使用亲本测序数据将杂合F1个体的测序数据分成两类,然后两类分别组装成两个亲本的单体型。该方法对拟南芥F1个体(杂合度1.36%)的组装结果显示,两个单体型的完成度和质量都达到较高水平。Triobin对来自亲本杂合区域的read分类效果较差,更适用于纯合亲本的情况。另外,家系测序的条件在很多研究中无法满足,限制了Triobin的应用范围。
遗传群体也是基因组分型的有力工具。通过遗传群体与不同测序技术结合,梨杂合基因组[42]和马铃薯杂合基因组[7]项目发表了组装杂合基因组完整单体型的方案。在杂合梨项目中,研究人员使用单倍体群体(12个花粉细胞)的测序数据对先前构建的3.8万个BAC进行分型,每条染色体的BAC被分成A、B两类,再分别组装成A、B单体型。为避免不同染色体互相干扰,分型前先用梨单倍体参考基因组识别BAC所属染色体,有效提高了分型的效率,但也限制了其在无参考物种上的应用效果。杂合马铃薯分型组装的流程包含3个阶段:(1)用高保真三代测序数据(HiFi read)组装出二倍体基因组的全部contig序列;(2)构建遗传图谱将contig分配到12个连锁群中,对应单倍体基因组的12条染色体;(3)同一连锁群的contig根据基因型分成两组,代表染色体的两个单体型。与其他分型方法类似,该流程也先区分不同染色体,再区分染色体的两个单体型。在阶段(2)中,研究人员开发了利用contig构建连锁群的方法,使用遗传连锁群区分不同染色体,避免了对已知参考基因组的依赖,扩展了应用范围。
高杂合基因组的组装和分型一直是基因组方法领域的难点,目前仍然没有相对简便的方法和工具。随着HiFi read 数据的应用以及hifiasm等综合利用三代测序、Hi-C数据优势的组装软件的开发[42-43],将促进杂合植物基因组解析的快速进展。
4 高重复基因组组装
重复序列在物种进化和功能调控中扮演不可或缺的角色,是基因组重要的组成部分。重复序列的序列相似性高、长度不一、拷贝数变化范围大,一直是组装中的难题。相比于二代测序技术,三代长读长测序可以跨过重复序列区域,提高重复序列的区分度,显著改善组装的连续性和重复序列组装的完整性、准确性,这种优势在85%的序列都来源于转座子扩增的玉米(Zea mays ssp. mays L.)基因组中得到充分体现。PacBio数据组装的玉米B73基因组,相对之前基于二代组装的版本,contig连续性提高了52倍,并且纠正了着丝粒区的组装错误,极大改善了基因功能区注释和转座子的进化分析[45-46]。
高重复序列基因组的另外一类代表是拥有巨大 基因组的植物,如火炬松(Pinus taeda L.,22 Gb, 82%)[47]、挪威云杉(Picea abies,20 Gb,>71%)[48]、 银杏(Ginkgo biloba,10 Gb,80%)[49],基因组70% 以上都是重复序列,远超拟南芥(20%)[50]、水稻(40%)[51]等模式植物。这些裸子植物都是杂合的,可以选择单倍的配子体胚乳进行测序。大型基因组的测序成本和组装技术难度都较大,最初发表的几个裸子植物基因组采用二代数据组装的方式,contig N50仅有几kb或者几十kb。近日阮珏团队利用~44× PacBio数据重新组装了银杏基因组[52],将contig N50由二代组装的48 kb[49]提高至1.58 Mb,并利用Hi-C挂载了12条染色体,是目前发表的最高质量的裸子植物基因组,也提高了研究人员对大型基因组的组装要求。2020年发表的大蒜(Allium sativum)基因组[53],经历3次全基因组复制及重复序列扩张,基因组达到16.9 Gb,其中91.3%都是重复序列,是迄今组装的重复序列比例最高的基因组,组装方法采用了 PacBio 构建contig、10×G文库连接成scaffold、最后用Hi-C数据挂载染色体。最新公布的杂合加州红杉(Sequoia sempervirens )基因组(6倍体,单倍体27 Gb)组装结果,研究人员使用PacBio HiFi数据和Hifiasm[42]软件获得47.47 Gb contig序列,N50达到1.92 Mb(https://downloads. pacbcloud. com/public/dataset/redwood2020/),并且组装时间仅需几天,展示了高准确率三代数据在大型植物基因组组装上的应用前景。
5 高倍性基因组组装
由于杂交和基因组加倍导致了多倍体植物的存在,一些重要的农作物例如小麦、棉花、马铃薯等都是多倍体,其基因组的解析是影响作物育种进展的重要因素。多倍体物种根据其形成机制分为异源多倍体和同源多倍体,异源多倍体中染色体来源于不同祖先,基因组内可以区分亚基因组,对组装干扰较少;而同源多倍体中多套染色体之间高度相似,相当于高杂合基因组,组装难度极大。
异源多倍体基因组通常可以当做纯合基因组进行组装,其重点是组装后区分亚基因组。国际小麦测序联盟解析六倍体栽培小麦(Triticum aestivum,AABBDD)基因组时利用流式细胞仪分离技术将21条染色体分离开,分别构建BAC文库进行测序和组装[54]。分离染色体的技术和成本要求较高,并不常见于普通植物研究。四倍体油菜基因组(Brassica napus,AACC)[55]和四倍体花生基因组(Arachis hypogaea,AABB)[56-57]的组装借助了二倍体祖先的测序数据区分出两个亚基因组。相对二代测序数据,三代测序数据可以更好区分相似序列,组装出连续性更长的contig,再结合全基因组遗传图谱或者Hi-C图谱区分异源染色体。2015年发表的四倍体棉花TM-1(Gossypium hirsutum,AADD)基因组由10万个BAC克隆和遗传图谱组装完成[58],2019和2020年发表的新版本的TM-1基因组均由PacBio数据和Hi-C图谱、光学图谱完成,提高了参考基因组质量,也提供了更高效、更低成本的多倍体组装 方法[59-60]。
相比异源多倍体由自然杂交产生,同源多倍体通过染色体加倍形成,遗传上多套染色体都可以联会,序列上同源区域相似度较高,在组装过程中互相干扰。在二代测序数据为主的时代,为构建物种的参考基因组,只能测序单倍体材料降低组装难度或者容忍、合并杂合区域。2017 年发表的六倍体甘薯基因组(Ipomoea batatas,B1B1B2B2B2B2)首次报道了同源多倍体植物的单倍体参考基因组和基因组30%区域的分型结果[61]。随后,2018年同源四倍体甘蔗基因组(Saccharum officinarum,1n=4x)首次攻克了同源多倍体单体型组装的难题[62],其关键步骤是使用BAC文库和三代测序数据克服序列相似性,组装出四倍体全部contig,再结合Hi-C图谱分成4套染色体。其中Hi-C分型软件ALLHIC[63]借助近缘物种高粱基因组,区分出甘蔗不同染色体的contig,再根据Hi-C互作信号对同源contig进行区分及锚定。同源四倍体紫花苜蓿(Medicago sativa L.,2n=4x)基因组的解析也使用了该方案,在二倍体苜蓿(M. truncatula)基因组的辅助下,成功获得了4套分型结果[8]。四倍体苜蓿首次使用了高准确率的PacBio HiFi数据进行多倍体组装,获得了比甘蔗基因组更好的contig 连续性。虽然同源多倍体的组装和分型在多个物种上都获得了成功,但是基于Hi-C的分型软件仍然要依赖单倍体的参考基因组,并且在处理差异较小的同源染色体时区分效果不明显,解析复杂同源多倍体基因组还需继续探索多种类型数据和技术整合[64]。
6 植物泛基因组进展
完成物种的参考基因组后,为挖掘和利用该物种的基因组资源,通常会进行群体重测序分析。传统分析方法是将个体的短序列匹配到参考基因组上识别个体间的差异。这种方式得到变异类型十分有限,尤其对于个体间遗传差异大的物种,单一参考基因组无法满足分析需求。泛基因组(pan-genome)通过对物种的不同个体进行测序及组装,尽可能地捕获该物种的全部遗传信息,为后续功能研究提供新的参考基因组,正逐渐成为挖掘物种遗传多样性的研究趋势。
泛基因组构建的方式有3种(图3)。早期研究由于测序数据较少,将个体测序数据比对到参考基因组,提取没有比对上的read进行组装,产生的新序列迭代补充到参考基因组上,这种方式称为迭代组装。2018年发表的3 010份水稻(Oryza sativa L.)测序是迭代组装构建泛基因组的经典研究[65],研究人员提出“map-to-pan”策略,从重测序个体中组装出268 Mb的非冗余序列,补充到日本晴参考基因组,作为后续变异检测和功能分析的基础。这种方式构建的泛基因组连续性较差,无法检测大的结构变异,重新组装的新序列也会导致泛基因组的冗余。
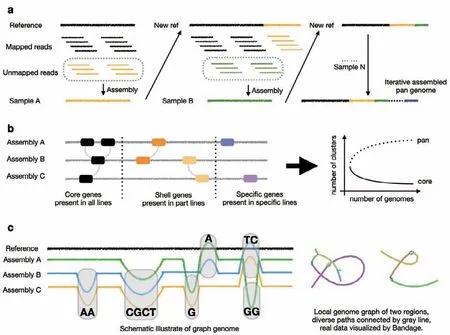
图3 泛基因组构建的三种方式Fig. 3 Three approaches of assembling pan-genome
第二种方式是从头组装个体基因组后再构建泛基因组。高质量的个体基因组是泛基因组分析的前提,因此组装成本较高。目前已发表的栽培稻-野生稻[66]、大麦(Hordeum vulgare L.)[67]和小麦泛基因组(Triticum aestivum L.)[68],均使用二代数据进行组装。其中大麦和小麦泛基因组中利用Hi-C数据将部分材料组装到染色体级别。从头组装有利于系统鉴定各类群的“存在-缺失”变异集,染色体水平的比较能够揭示全基因组大规模序列重排和结构变异,为解析复杂表型的遗传机制提供更精确的信息。大麦泛基因组的分析揭示了不同种质中两个高频染色体异位与育种和驯化的关系,展示了组装的质量决定泛基因组分析的精度以及其在育种中的应用。
第三种方式是近年来快速发展的图基因组(graph-based genome)[69-70],用图上的路径(path)表示不同个体中相同和差异的序列。图参考基因组的构建一般基于从头组装的基因组,将不同个体的基因组比对到线性参考基因组提取变异,所有个体的变异经过去冗余,再与线性基因组进行整合,通过多条路径的方式展示各种变异。图基因组考虑了个体间的相似性和差异性,也能更加直观的展示群体中复杂的结构变异。图基因组相对线性基因组,能够更好的协调多个基因组的坐标对应关系,以最小的数据结构保留全部个体的序列信息,将在泛基因组分析模型中获得广泛应用[71]。2020年发表的大豆(Glycine)泛基因组[72],是第一个构建高质量的图参考基因组的作物,研究人员使用第三代测序数据从头组装选择了26个代表性材料并锚定到染色体,平均contig N50达到了22.6 Mb,获得了在泛基因组分析中最高的组装连续性,对后续的泛基因组研究的提出了更高的标准[73]。
7 测序技术发展与组装质量
早期使用Sanger测序BAC等大片段克隆,再将大片段拼接成基因组。人类基因组项目用该方法完成了大肠杆菌、酵母、线虫及果蝇等模式物种的标准参考基因组。此方法完成的基因组质量较好,但是成本过高,无法满足日益增长的物种组装需求。
在过去十几年间,第二代测序技术快速发展,每Gb数据价格降低到50元以内,片段长度从30 bp提高到300 bp,并保证了较高的碱基准确率(>99%),使完成的基因组数量得到迅速增长。二代测序建库过程中需要PCR扩增,存在GC偏好性,有些区域无法被二代测序覆盖,影响组装完整性。由于二代测序的读长较短,通常构建2 kb-40 kb的mate-paire文库以跨过重复序列等难组装区域。这导致用二代数据完成的基因组含有大量gap,contig通常只有几十kb。此外,读长限制使二代测序难以解决基因组复杂区域,如着丝粒,端粒等富含串联重复片段区域,组装结果远达不到基因组“完成图”的质量。
以PacBio和Nanopore为代表的第三代测序技术无需PCR建库过程对基因组覆盖更均匀,实现了单分子测序,读长可以达到几十kb到上百kb。Nanopore ultra-long 测序技术,甚至可以产生Mb级别的read[74-75]。长度优势使第三代long read能够跨过长距离复杂区域,提供足够多标记区分相似、同源片段,将组装contig N50提高到Mb甚至几十Mb级别。Pacbio和Nanopore已经成为基因组组装最常用的数据类型,许多之前使用二代数据组装的物种基因组,使用三代数据重新进行了组装,提高contig连续性并且补充之前二代测序没有覆盖的区域。
PacBio的CLR(continuous long reads)数据原始碱基准确率为85%-92%。碱基错误是随机的,增加测序深度进行校正后,一致性序列准确性可以达到99.99%。Nanopore 数据的原始碱基准确率与CLR相似,但是错误不完全随机,纠错后准确率可以提高到99%。然而,在植物杂合基因组或者高重复序列基因组中,同源或者多拷贝的序列之间差异只有1%-2%,远低于三代序列的测序错误(10%-15%),对原始数据进行纠错不可避免会合并基因组上的相似序列,在后续组装和分型过程中损失该类序列的信息。在使用CANU等软件组装这类基因组时,有时纠错阶段会将原始数据量减少至三分之一,导致最后组装结果远小于预估基因组大小。并且原始数据纠错耗时较长,在大型基因组(>10 Gb)组装中成为短板因素。
近两年来PacBio公司推出的高保真HiFi read,碱基准确率>99%。HiFi数据的高准确率显著提高了参考基因组组装的质量并且精减了原始序列纠错、组装结果抛光等步骤,是当前质量认可度最高的测序数据。HiFi read测序时对DNA插入片段进行多次循环读取,以牺牲长度换取高准确率, 平均读长只有CLR的1/2(10-20 kb vs 20-40 kb),并且通量只有CLR的1/5,当前一张SMRT cell芯片可以产出>100 Gb CLR read数据,而只能产出20-25 Gb HiFi read数据,无法跨过长距离复杂区域,且数据有效率较低、成本较高,这些是HiFi数据在解决大型、复杂基因组时的局限。
基因组组装的质量在很大程度上取决于测序技术产出的片段长度和准确率。HiFi提供了高精度单分子测序,Nanopore ultra-long提供了超长片段,相信在不久的将来随着测序成本的降低,这两种技术的综合应用能推动植物基因组进入端粒到端粒的“完成图”组装时代。
8 总结与展望
当前各种建库技术、测序平台都在不停发展,获得高质量、高深度、多维度测序数据的成本在快速降低,构建研究物种的参考基因组成为基因组项目的常规任务。随着被解析的基因组数量增加,植物基因组的易变性和多样性不断在各种复杂基因组中得到体现。在实际研究中,每个待组装的基因组所面临的技术问题和后续的分析需求也不尽相同。在项目初期做好基因组特征评估和对组装质量的预期,再选择测序和组装策略是比较明智的做法。
泛基因组正逐渐成为研究作物驯化与基因功能的新参考基因组。越来越多的个体基因组完成了高质量组装,使泛基因组的构建更具有实用价值,同时也对数据的存储、可视化以及流程化分析提出新的要求。如何整合参考基因组与多组学数据,实现泛基因组指导作物育种改良成为新的热点方向。
除组装方法外,植物基因组面临的还有测序技术方面的挑战。由于细胞壁的存在,以及大量的多糖、次生代谢产物等,从植物细胞中提取高质量、大片段DNA构建测序文库经常会遇到困难。例如Nanopore ultra-long 测序技术受限于植物DNA提取技术,目前仍未在植物基因组中有应用报道。未来相关测序实验技术的突破也能够为复杂植物基因组解析提供新的契机。
猜你喜欢
基层中医药(2022年4期)2022-07-22
山西农业科学(2021年3期)2021-12-06
汉字汉语研究(2021年2期)2021-08-30
种子(2021年3期)2021-04-12
今日农业(2020年14期)2020-12-14
诊断学(理论与实践)(2020年6期)2020-12-09
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
