
Schweninger-Buzzi型原发性皮肤松弛症1例
2021-08-02 02:13韩凤娴李敬梁颖于霖
中国中西医结合皮肤性病学杂志 2021年3期
韩凤娴,李敬,梁颖,于霖
(天津市中医药研究院附属医院,天津 300120)
皮肤松驰症[1]是1892年Jadassohn描述并报道了此病。临床上将其分为原发性皮肤松弛症(PA)(发病前皮损部位为正常皮肤或只有轻度非特异性炎性反应过程),继发性皮肤松弛症(SA)[发病前皮损部位存在其他皮肤病,临床常见的如痤疮、水痘、系统性红斑狼疮(SLE)、梅毒等]。PA又可分为3型:①Jadassohn-Pellizari型皮肤松弛症,又称红斑性皮肤松弛症,其特点为在皮肤发生萎缩之前局部先有红斑、风团等炎性反应改变。②Schweninger-Buzzi型皮肤松弛症,又称无红斑性皮肤松弛症,其特点为临床和组织病理变化始终缺乏炎性反应。③皮肤痘疮样斑状萎缩。较常见的为Jadassohn-Pellizari型皮肤松弛症。
1 临床资料
患者女,24岁。颈、躯干、四肢散在丘疹、斑块,隆起呈疝囊状,无自觉症状8年,于2018年5月来我院门诊就诊。患者8年前偶然发现颈、躯干、四肢散在十余个绿豆至花生大、肤色圆形、椭圆形境界清楚的丘疹、斑块,质地柔软,无痛痒,皮疹自发现后无明显变化,亦无新发皮疹。曾就诊于多家医院,未予确诊及治疗。患者既往体键,家庭成员中无相同或类似疾病患者。个人史及家族史无特殊。
体格检查:各系统检查未见异常。皮肤科检查:颈后,左腋前方,腰部,四肢散在十余个大小不一的圆形、椭圆形柔软性丘疹、斑块,直径0.5~1.5 cm,表面皮肤正常,无红斑水肿及毛细血管扩张,触之有疝囊样感觉,用手指按触时可凹下似袋状。肢体伸展或皮肤绷紧时皮疹可消失或不易发现,见图1。皮损组织病理检查:表皮与真皮萎缩,真皮血管周围轻度炎性细胞浸润,真皮弹力纤维染色显示正常的弹力纤维消失,见图2。
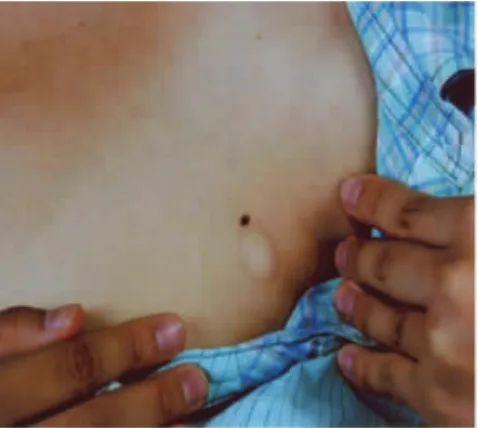
图1 患者皮损临床照片
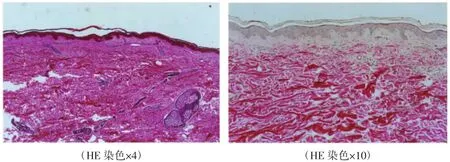
图2 皮损部位组织病理图片
根据患者的病史、临床表现、病理检查结果,临床诊断为Schweninger-Buzzi型皮肤松弛症。
2 讨论
皮肤松弛症临床特点为在正常皮肤上出现境界清楚的柔软松弛的丘疹或斑块,可以轻度凹陷,或呈袋状隆起,指尖触诊感觉皮肤缺乏弹性,有嵌入疝空的感觉。皮疹直径0.5~2 cm左右,颜色可以是皮色、白色或棕灰色,皮疹好发于躯干及四肢近端,头颈部较少见,四肢远端、手足掌及黏膜部位更为罕见。病程缓慢,部分皮损可自然消退,或发展至一定程度可终身不变,患者无任何自觉症状。女性发病率高于男性。家族多发病例常染色体显性和隐性发病均有报道[2]。
组织病理上常规染色与正常皮肤无明显区别,真皮内可或多或少见到轻度炎性细胞浸润包括淋巴细胞、浆细胞、组织细胞,弹力纤维染色可见真皮网状层弹力纤维几乎完全丧失。电镜下可见颗粒状的弹力纤维变性和残余的成片段的细小弹力纤维[3]。皮损直接免疫荧光显示有非特异性的免疫复合物沉积。
该病的发病机制尚不清楚,弹力纤维的降解可能与一种金属蛋白酶及其抑制剂失衡有关,也可能与吞噬细胞对弹力纤维的吞噬活性增加有关,Zaki等[4]证实在电镜下PA患者真皮内巨噬细胞对弹力纤维的吞噬活性明显增强。Hodak等[5]报道几例皮肤松弛症患者皮损直接免疫荧光可见真皮表皮交界处IgM、C3、C1q、IgA、IgG呈颗粒状沉积。组织切片中浸润的细胞以CD4+T辅助细胞为主,说明发病过程中存在免疫炎性反应。一些病例报道皮肤松弛症患者同时合并抗磷脂抗体综合征[6]、人类免疫缺陷病毒(HIV)感染[7]、SLE[8]、自身免疫性溶血、硬皮病、低补体血症等自身免疫性疾病。近年来发现,皮肤松弛症患者大多伴有抗磷脂抗体异常(合并或不合并抗磷脂综合症)。抗磷脂抗体如何引起弹力纤维降解尚不清楚,有学者因为在病损部位发现微血栓而猜想可能因为微血栓引起缺血进而导致弹力纤维变性。Jeong等[8]提出弹力纤维和磷脂可能有相似的抗原表位,使弹力纤维成为抗磷脂抗体的潜在靶组织最后受到攻击。这些暗示自身免疫反应可能与皮肤松弛症的发病有关。越来越多的学者认为皮肤松弛症不是原发或自发的,而是自身免疫性疾病病谱中的一种皮肤症状,有必要对原发患者进行系统的免疫抗体检查并长期随访这样不仅可以发现潜在的血栓风险又可以更全面的了解疾病[9-10]。
本例患者于正常皮肤上发生皮损属于Schweninger-Buzzi型皮肤松弛症,为临床少见一型。本病需与神经纤维瘤病、真皮中层弹性组织溶解症及结缔组织痣等疾病相鉴别。有尝试秋水仙碱、氨苯砜、液氮,糖皮质激素等治疗效果均不明显,对已形成的皮肤松弛性皮损尚无确切有效的治疗方法,对影响美观的皮损可行手术切除。对原有疾病的控制可以减少新的皮损出现。
猜你喜欢
西部皮革(2022年19期)2022-11-20
西部皮革(2022年21期)2022-11-16
乳业科学与技术(2022年2期)2022-04-15
中国油脂(2022年1期)2022-02-12
农产品加工(2021年8期)2021-05-20
中华养生保健(2020年1期)2020-11-16
家庭医药·快乐养生(2019年2期)2019-03-04
汽车与驾驶维修(维修版)(2018年8期)2018-09-21
大陆桥视野·下(2017年8期)2017-09-19
医学研究杂志(2015年9期)2015-07-01
