
基于图像分析的玉米抗拟轮枝镰孢穗腐病的QTL定位
2021-07-21 10:40闻竞沈彦岐王梓钰李世界莫蓝月雷宇豪张艳韩四平
中国农业科学 2021年13期
闻竞,沈彦岐,王梓钰,李世界,莫蓝月,雷宇豪,张艳,韩四平
基于图像分析的玉米抗拟轮枝镰孢穗腐病的QTL定位
闻竞,沈彦岐,王梓钰,李世界,莫蓝月,雷宇豪,张艳,韩四平
吉林省农业科学院农业生物技术研究所,长春 130033
【】玉米穗腐病是一种在全世界广泛发生且危害严重的真菌性病害,其中,拟轮枝镰孢引起的穗腐病(ear rot,FER)在中国发生最为普遍。通过图像分析方法进行FER抗病QTL定位,并对前期通过病害评级方法定位的FER抗病QTL进行验证,探索一种新的玉米穗腐病的病害鉴定方法,为玉米穗腐病的遗传改良提供依据。利用高感FER的自交系(ZW18)分别与3个高抗自交系(承351、丹598和吉V203)构建F2群体(F2-C、F2-D和F2-J)和相应的F2﹕3家系,通过图像分析的方法获得每个F2﹕3家系的果穗病斑百分比,进而定位玉米FER抗病QTL。3个群体共定位到18个FER抗病QTL,其中,分别位于2.02—2.03 bins、4.06—4.07 bins和8.06 bin上的3个QTL(、和)可解释的表型变异率分别高达21.80%、25.80%和27.40%。F2-J群体的与F2-C群体的和F2-D群体的在第1染色体均有重叠区间,可解释的表型变异率达到16.50%。来自F2-D群体的与F2-J群体的在第8染色体8.05 bin有重叠区间,且抗性基因均来源于抗性亲本。F2-C群体的与F2-J群体的在第4染色体4.06—4.07 bins有重叠区间。另外,与之前通过病害评级方法定位的结果相比,、和在1.06—1.07 bins与评级方法定位的抗病位点重合,和5在4.06—4.07 bins与评级方法定位的抗病位点和重叠,与在2.04 bin的定位区间完全重合,与在S2重复中定位到9.03—9.05 bins的重叠区间,且来源于相同的抗源。定位到18个FER抗性位点,其中,位于1.04—1.07 bins、4.06—4.07 bins和8.05 bin上的抗病位点在不同群体中均可以被检测到,位于2.04 bin和9.03—9.05 bins上的抗病位点用不同的检测方法可以被检测到,表明在这些区间可能存在FER的抗性位点。QTL的定位区间在不同群体中的重叠性在一定程度上验证了定位区间的真实性,不同方法之间定位到重叠区间,说明利用图像分析方法定位FER抗病QTL具有一定的准确性。
玉米;拟轮枝镰孢穗腐病;抗性;QTL
0 引言
【研究意义】玉米是粮饲兼用作物,又是重要的工业原料,在中国粮食安全保障体系中占有举足轻重的地位。病虫害是中国玉米生产的主要限制因子,每年造成的产量损失占总产的10%以上,严重威胁到中国的粮食安全。玉米穗腐病是一种在全世界广泛发生且危害严重的真菌性病害[1-4],也是中国各个玉米产区的主要病害。一般年份玉米穗腐病的发病率为10%—20%,严重年份可达30%—40%,感病品种的发病率高达50%左右[5],而且其发病程度呈逐年加重的趋势。玉米穗腐病不仅会引起果穗腐烂而导致直接减产,而且有些病原菌还会在被侵染的籽粒中产生大量的有害毒素,严重影响畜牧业生产,进而威胁人类的身体健康和生命安全[6-7]。因此,定位玉米抗穗腐病主效QTL、发展分子标记,为抗病分子育种服务,无论在抗病理论探索还是育种实践应用上都具有重要的意义。【前人研究进展】玉米穗腐病的致病菌种类繁多,国内外报道有40多种,这些病原菌可以单独或者复合侵染引起发病[8-9]。目前已报道的玉米穗腐病的主要致病菌包括镰孢菌(spp.)、木霉菌(spp.)、青霉菌(spp.)、曲霉菌(spp.)、平脐蠕孢菌(spp.)和链格孢菌(spp.)等[10-12]。不同国家及地区玉米穗腐病病原菌种类及优势种类差异明显,国内外报道不尽一致,而且不同区域环境下,穗腐病的主要致病菌存在差异[13-15]。已有研究结果表明,国内玉米穗腐病的病原菌主要以拟轮枝镰孢()和禾谷镰孢()为优势种,分别引起拟轮枝镰孢穗腐病(ear rot,FER)和禾谷镰孢穗腐病(ear rot,GER),其中,FER在中国发生最为普遍[16-21]。目前,国内外已利用多种方法开展对FER抗病QTL的定位研究[22-30]。有研究利用重组自交系进行QTL定位,定位到的抗性QTL位于第3、4、5、6染色体[31]。也有研究结果显示在3.04 bin可能含有多个抗病基因[32]。通过全基因组关联分析,在第4染色体上定位到了FER的抗病QTL,表型变异率达18%[33]。也有研究通过全基因组关联分析与基因组预测相结合的方法,将FER的抗病QTL定位在第3、7、9、10染色体上[34]。【本研究切入点】现有的针对玉米穗腐病的防治措施(如喷施杀菌剂、翻耕、间混作等化学防治和耕作措施)效果均不理想[35]。另外,玉米籽粒直收已成为玉米育种的方向,而籽粒直收过程中,无法对发生穗腐病的果穗进行剔除。机械化收获的不断推进,将会对品种穗腐病抗病方面的要求变得更为严苛。因此,培育和推广抗病品种成为目前亟需解决的问题。然而,目前还没有可以直接用于分子育种的抗穗腐病遗传稳定的QTL或者基因被报道。【拟解决的关键问题】本研究通过借助图像处理软件计算果穗病斑面积的百分率获得群体的病害数据进行玉米FER抗病QTL的定位,探索一种新的玉米穗腐病抗性鉴定方法,同时对前期通过病害评级方法定位的FER抗病QTL进行验证。
1 材料与方法
1.1 试验材料
植物材料:高感FER的自交系ZW18分别与高抗FER的玉米自交系承351、丹598和吉V203构建3个F2群体(F2-C、F2-D和F2-J)和F2﹕3家系,F2群体大小分别为124、200和176(图1)。前期已分别构建3个F2群体的遗传连锁图谱,总遗传距离分别为2 754.2、1 862和2 086.1 cM,平均每两个相邻标记之间的遗传距离分别为5.91、4.22和3.75 cM。在构建遗传连锁图谱时将感病亲本的基因型设为“1”,抗病亲本的基因型设为“2”。F2群体的F2﹕3家系用于玉米FER的病害鉴定。
病原菌:拟轮枝镰孢()由中国农业大学徐明良教授提供。
1.2 试验方法
1.2.1 田间设计 将亲本自交系和3个F2群体的F2﹕3家系分别种植于吉林省公主岭地区和松原地区2个试验点,每个点设置2个重复。每份材料的每个重复种植1行,行长5 m,行距0.6 m,株距0.25 m,每行种植21株。
1.2.2 病原菌的培养 将拟轮枝镰孢菌接种到PDA培养基平板上,在培养箱中28℃培养约1周,待菌丝长满平板,4℃保存待用。取40 g绿豆加入适量水煮沸20 min,过滤后加入20 g蔗糖,1 g KH2PO4,定容至1 L,121℃高压蒸汽灭菌30 min,冷却后4℃保存备用。将1/2板已培养好的带菌平板中的培养基切成0.25 cm2的小块放入500 mL 5%绿豆煎汁液体培养基中,28℃ 150 r/min震荡培养2 d,过滤菌丝,收集孢子置于4℃保存备用。在接种前一天,用无菌蒸馏水配制成浓度为5×106个/mL的孢子悬浮液。在孢子悬浮液中加入表面活性剂吐温-80,吐温-80在孢子悬浮液中浓度为2 µl·mL-1,混匀。

A:承351;B:丹598;C:吉V203;D:ZW18 A: Cheng351; B: Dan598; C: JiV203; D: ZW18
1.2.3 人工接种 根据往年经验,接种的最佳时期为玉米吐丝后12 d,用注射器在果穗中部注射2 mL孢子悬浮液,接菌后用手指轻捏伤口处,防止菌液外流。
1.2.4 病害鉴定 在玉米成熟期(吉林省公主岭和松原9月下旬)进行田间发病调查。将各接种处理的玉米果穗剥去苞叶,并对每个重复中的每个F2﹕3家系的所有果穗发病最严重的部位拍照,拍照设置比例尺,保证每张图片中玉米籽粒的大小一致,然后利用Photoshop软件处理图片。首先利用磁性套索工具将玉米果穗从图片中分离,利用魔棒工具选中病斑位置,并用绿色填充;随后选择反向,选中果穗的未发病部位,将其填充为红色。分别选中果穗发病和未发病的部位,点击“分析”,选择“数据测量”,记录测量结果中各部分的面积大小,计算每个重复中每个F2﹕3家系全部玉米果穗的发病面积占总面积的百分比(图2)。以每个F2﹕3家系中所有个体的病害鉴定值的均值作为对应的F2个体的病害鉴定值。
1.2.5 方差分析方法 利用R软件进行表型的方差分析,采用=μ+α+β+()+εGLR的模型。其中:α表示基因型的效应,β表示环境的效应,()表示基因型和环境的互作效应,表示残差效应。
1.2.6 QTL初定位方法 采用F2群体进行玉米抗穗腐病QTL的初定位。表型数据采用4个重复(G1、G2、S1、S2)、每个地点2个重复的平均值(G、S)和最佳线性无偏估计值(best linear unbiased estimation,BLUE)的数据进行计算。结合F2群体的遗传连锁数据和表型数据利用WinQTLCart软件[36]采用复合区间作图法(composite interval mapping,CIM)[37]进行玉米抗穗腐病QTL的扫描,LOD值的阈值采用排列测验的方法由软件自动产生,其中排列测验的次数为300,显著水平为0.05。

A:原图;B:去除背景的玉米果穗图片;C:用不同颜色标注病斑和未发病部位的图片;D:数据分析
2 结果
2.1 表型分析
对定位群体的材料进行玉米穗腐病抗性的鉴定。人工接种拟轮枝镰孢菌后,在公主岭的第1个重复(G1)中亲本自交系承351、丹598、吉V203和ZW18的发病面积百分比分别为0、0、0和49.22%;在公主岭的第2个重复(G2)中亲本自交系承351、丹598、吉V203和ZW18的发病面积百分比分别为0、0、0和82.26%;在松原的第1个重复(S1)中亲本自交系承351、丹598、吉V203和ZW18的发病面积百分比分别为0、0、1.30%和63.85%,在松原的第2个重复(S2)中亲本自交系承351、丹598、吉V203和ZW18的发病面积百分比分别为0、2.40%、0和55.38%。在F2群体中,对玉米抗穗腐病的抗性没有呈现双峰的分布(图3),说明玉米对穗腐病的抗性不是由单基因控制的,而是由数量抗性位点控制的。
2.2 玉米穗腐病抗性的方差分析
对由抗病亲本承351、丹598和吉V203分别与感病亲本ZW18构建的F2群体(F2-C、F2-D和F2-J)的不同重复进行方差分析,结果表明,每个群体中不同重复间的发病水平基本相当。抗病亲本(承351、丹598和吉V203)在不同的地点不同重复中均表现一定的抗性,感病亲本ZW18均表现明显感病。根据F2-C、F2-D和F2-J群体在不同重复的表型数据估算的FER抗性遗传力分别为0.63、0.29和0.56(表1)。方差分析结果表明,在F2-C群体中,不同基因型之间和不同地点之间的FER抗性存在显著性差异,另外,基因型与环境之间的互作效应显著。在F2-D群体和F2-J群体中,不同基因型之间和不同地点之间的FER抗性存在显著性差异(表2)。另外,3个F2群体的残差的偏度分析结果表明残差符合正态分布(表2和图4)。
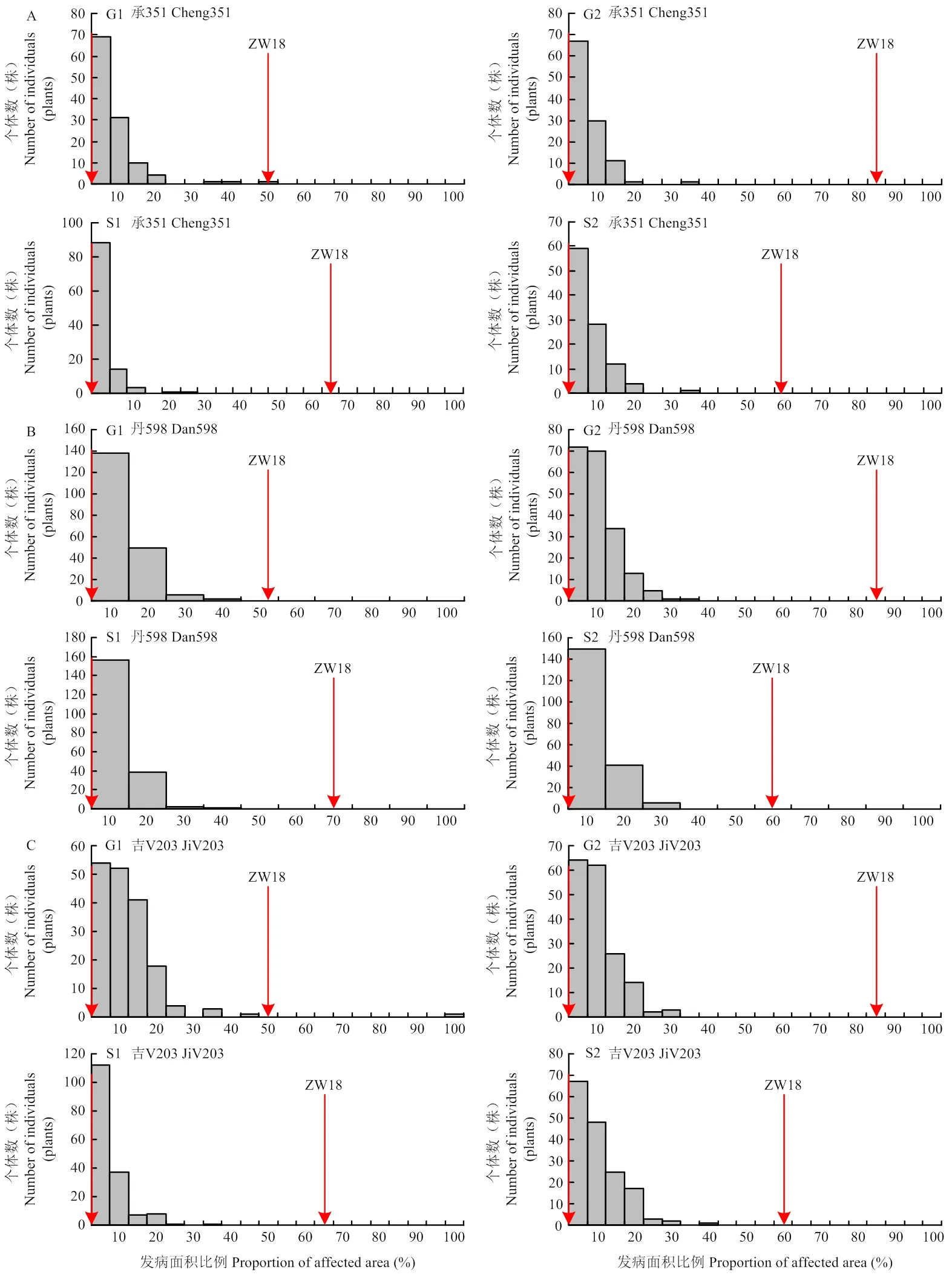
A:F2-C;B:F2-D;C:F2-J。G1:公主岭第1个重复;G2:公主岭第2个重复;S1:松原第1个重复;S2:松原第2个重复。下同

表1 亲本及F2群体的发病面积百分比分析结果
G1:公主岭第1个重复;G2:公主岭第2个重复;S1:松原第1个重复;S2:松原第2个重复。下同
G1: 1streplicate in Gongzhuling; G2: 2ndreplicate in Gongzhuling; S1: 1streplicate in Songyuan; S2: 2ndreplicate in Songyuan. The same as below
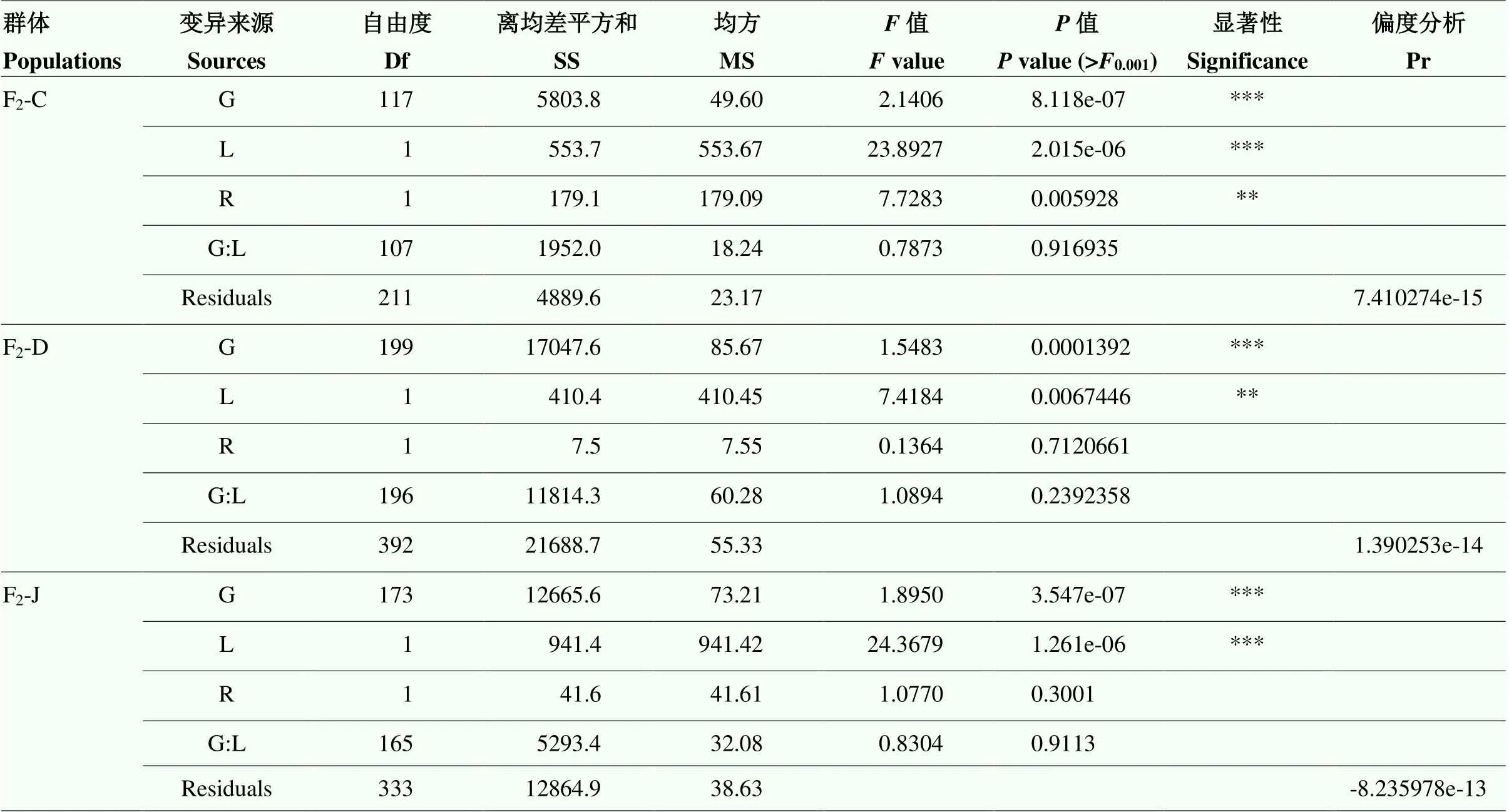
表2 人工接种拟轮枝镰孢菌后玉米穗腐病抗性的方差分析
G:基因型;L:地点;R:重复;G:L:基因型与环境间的互作;Residuals:残差效应
G: Genotype; L: Location; R: Replicate; G:L: Interaction between genotype and environment; Residuals: Residual effect

A:F2-C;B:F2-D;C:F2-J
2.3 玉米FER抗病QTL的定位
利用3个F2群体(F2-C、F2-D和F2-J)对玉米FER抗病QTL进行了定位,WinQTLCart经过300次排列测验之后生成相应的LOD阈值,共定位到18个抗性QTL。利用F2-C群体共定位到5个抗性QTL,分别位于1.04—1.06 bins、2.02—2.03 bins,4.06—4.07 bins、8.06 bin和8.07—8.08 bins。其中、、和的抗性等位基因来源于抗病亲本承351,的表型变异率最大,为26.00%—27.40%;的抗性等位基因来源于感病亲本ZW18,解释了7.1%的表型变异率(图5-A和表3)。利用F2-D群体定位到5个抗性QTL,分别位于1.07 bin、2.04 bin、3.03—3.04 bins、8.05—8.06 bins和10.06 bin。其中、、和的抗性等位基因来源于抗病亲本丹598,单个QTL的表型变异率为7.94%—12.32%;的抗性等位基因来源于感病亲本ZW18,表型变异率为10.82%(图5-B和表3)。利用F2-J群体定位到8个抗性QTL,分别位于1.06—1.07 bins、3.04—3.05 bins、3.06—3.07 bins、4.01—4.03 bins、4.05—4.07 bins、8.03—8.05 bins、9.03—9.05 bins和9.06—9.07 bins。其中、、和的抗性等位基因来源于抗病亲本吉V203,的表型变异率最大,达到17.96%;、、和的抗性等位基因来源于感病亲本ZW18,表型变异率最大的为,达到13.46%(图5-C和表3)。另外,已定位到的18个QTL由加性效应和显性效应共同遗传(表3)。
3个F2群体在第1和第8染色体均定位到抗性QTL。其中,F2-C群体的与F2-J群体的在1.06 bin(e1178476—e1188929)有重叠区间,F2-D群体的与F2-J群体的在1.07 bin(e1197737—e1200620)区间重叠。这3个QTL除的抗性等位基因来源于感病亲本ZW18外,与的抗性等位基因均来源于抗性亲本。F2-C群体的在BLUE中被定位,F2-J群体的在S2重复中被定位。与和均有重叠区间,可解释的表型变异率达到16.50%,说明可能是FER的抗病“热点”。另外,3个F2群体在第8染色体上均有定位区间。其中,来自F2-D群体的与F2-J群体的在8.05 bin(e8130926—e8144487)有重叠区间。在BLUE中被定位,表型变异率为10.85%;在S重复中被定位,表型变异率为8.15%。二者的抗性等位基因均来源于抗性亲本。此外,F2-C群体的与F2-J群体的在第4染色体4.06—4.07 bins(e4168742—e4171651)有重叠区间。在G1和BLUE中被定位,可解释的表型变异率分别为25.80%和19.60%;在G2和S2中被定位,可解释的表型变异率分别为17.96%和8.16%。
前期利用病害评级的方法进行抗病QTL定位,共挖掘到20个抗病位点,其中,位于1.03 bin、1.04—1.07 bins、2.04—2.08 bins、4.05—4.08 bins、6.03—6.06 bins和7.01—7.02 bins上的6个位点在不同的群体中都能检测到,说明这6个位点可能是拟轮枝镰孢穗腐病的抗病“热点”[38]。利用图像分析的方法共定位到18个抗病QTL,其中,、和在1.06—1.07 bins与评级方法定位的抗病位点重合(表4)。在S1和S2同时被定位到,在S2中的表型变异率为16.73%,同样在S2群体中定位到,表型变异率为16.50%,二者的表型变异率极为接近,且抗性等位基因均来源于抗性亲本吉V203,均受加性效应的影响。此外,利用图像分析的方法定位到的和5在4.06—4.07 bins与评级方法定位的抗病位点和重叠[38](表4)。和5在G1、G2、S2和BLUE重复中被定位,可解释的表型变异率为8.16%—25.8%;和在G1、G2、G和BLUE重复中被定位,可解释的表型变异率为12.02%—21.81%。2种方法定位到的位点表型变异率差异不大,抗性等位基因均来源于抗性亲本承351和吉V203,均受加性效应的影响。与在2.04 bin的定位区间完全重合,的表型变异率为7.94%,的表型变异率为7.02%,二者的表型变异率差异不大,且均在BLUE重复中定位到,抗性等位基因均来源于抗性亲本丹598,受加性效应的影响[38](表4)。另外,与也同时在S2重复中定位到9.03—9.05 bins的重叠区间,二者的表型变异率分别为3.61%和4.10%,抗性等位基因均来源于感病亲本ZW18[38](表4)。利用图像分析的方法定位到的抗性位点与病害评级法定位的QTL有重叠区间,说明该方法较为可靠,定位结果的准确性较高。、、、和定位到的抗性QTL用不同的方法均可定位到,说明这5个位点可能是拟轮枝镰孢穗腐病的抗病“热点”。
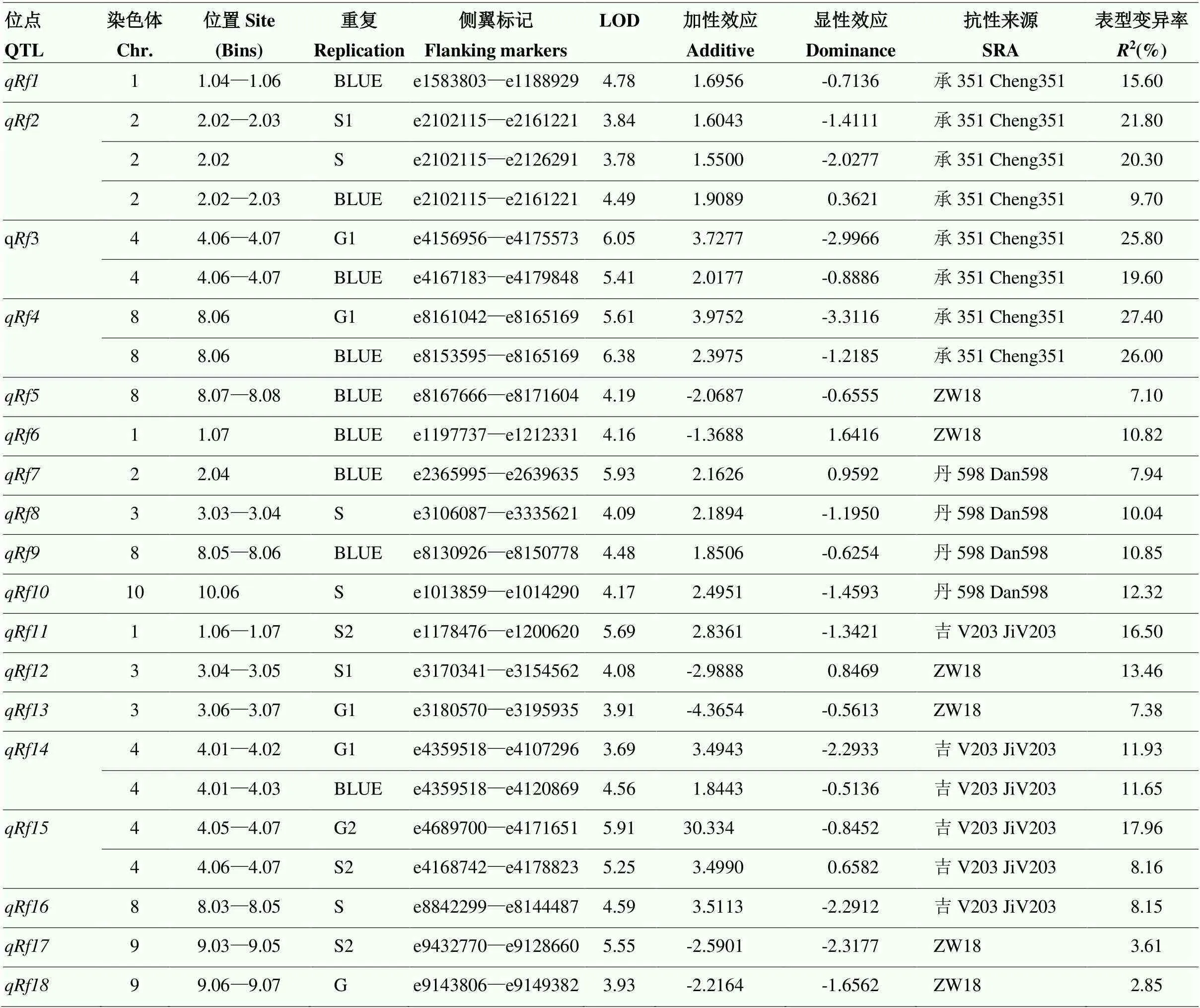
表3 玉米FER抗病QTLs及其参数
G:G1和G2的平均值;S:S1和S2的平均值;BLUE:最佳线性无偏估计值。下同
G: Average of G1 and G2; S: Average of S1 and S2; BLUE: Best linear unbiased estimation. The same as below

A:F2-C;B:F2-D;C:F2-J。G:G1和G2的平均值;S:S1和S2的平均值;BLUE:最佳线性无偏估计值
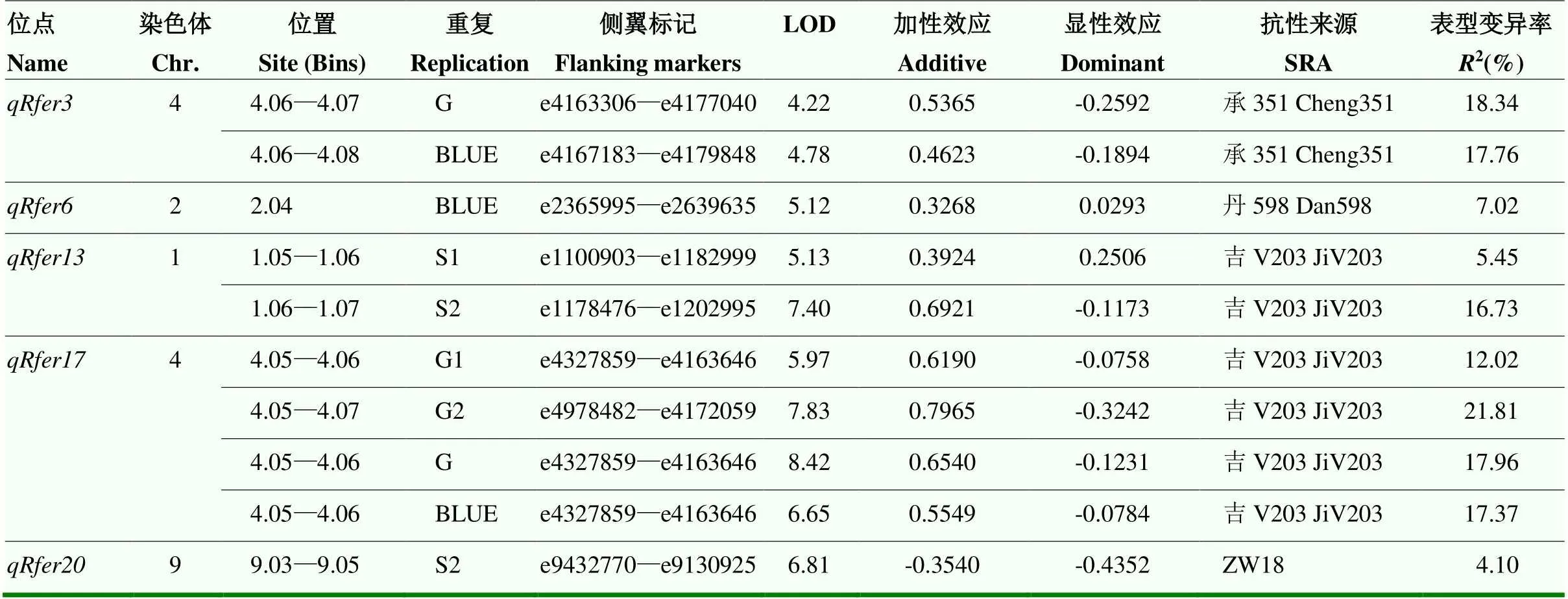
表4 利用评级法分析的玉米FER抗病QTL及其参数
3 讨论
在以往的玉米穗腐病病害调查中,大多采用的是病情分级的方式(《玉米抗病虫性鉴定技术规范,NY/T 1248.8—2016》)。该种鉴定方法是根据病斑所占的百分比进行人为评级,存在一定的主观因素,而且同一评级的发病面积占果穗总面积的比例的范围较大[39-40]。以9级为例,发病面积占果穗总面积的51%—100%均定为9级,同一等级的果穗发病面积百分比可能会有较大差异。因此,单从评级水平无法精确评价果穗的真实发病情况。为了探索一种新的玉米FER的病害鉴定方法,本研究采用对发病果穗的图片通过软件分析并计算出发病面积所占的百分比的方法获取病害数据。该种方法虽然工作量相对较大,耗费时间较长,操作较为繁琐,但可以精确计算不同果穗的发病面积所占的百分比。
本研究共定位到18个FER抗性位点,其中位于1.04—1.07 bins、4.06—4.07 bins和8.05 bin上的抗病位点在不同的群体中均可以检测到,位于2.04 bin和9.03—9.05 bins上的抗病位点用不同的检测方法可以检测到,说明在这些区间可能会存在FER的抗性位点。目前,国内外已有一些关于玉米穗腐病有效QTL位点的研究报道。有研究以BT-1和N6为亲本构建了250个重组自交家系材料,定位到的抗性QTL位于4.06 bin,本研究也得到了相同的结果[31]。此外,有研究结果表明在3.04—3.05 bins、9.04 bin、9.05 bin、9.06—9.07 bins均有QTL与FER抗性显著相关,这些区域与本研究中、和的定位区间重叠[22-28,33]。但也有研究结果显示定位到的FER抗性QTL在1.01 bin、5.05 bin、5.08 bin、5.09 bin、7.02—7.04 bins、9.00—9.01 bins和10.03—10.04 bins,本研究并未找到与之重叠的QTL[23-25,29-30,32-34,41]。与前期利用病害评级的方法进行的抗病QTL的定位结果相比,有多个QTL的定位区间重叠,、、、和分别与之前通过病害评级的方法定位到的、、、和20的定位区间重叠,而且来源于相同的抗源[38]。以上QTL的定位区间的重叠性在一定程度上验证了其真实性,但是还需要通过精细定位和遗传效应分析进一步验证以上QTL对玉米FER的抗病功能。在2种定位方法中有未能同时定位到的QTL,可能是由于2种方法在发病情况的表型鉴定方面存在差异,从而造成定位偏差。在本研究中定位到的18个QTL中,来源于抗病亲本承351的、和可解释的表型变异率分别可高达21.80%、25.80%和27.40%,而且可在不同处理的表型数据的定位中检测到。后期对、和的精细定位、遗传效应分析以及功能基因的克隆将对玉米FER的抗病遗传改良提供重要的遗传资源和理论参考。然而,在本研究中并未定位到可以在所有不同处理的表型数据(G1、G2、S1、S2、G、S和BLUE)中均能定位到的QTL。根据前期报道,玉米穗腐病的遗传机制非常复杂[42],而且发病对环境条件非常敏感[43-44],所以很难在不同的环境条件下获得较为一致的群体病害数据,从而影响对抗病QTL的定位结果。因此,对于玉米穗腐病这样的复杂的数量性状,可通过与不同的方法的结合进行功能位点挖掘,比如全基因组关联分析,转录组分析、代谢组分析等。
4 结论
共定位到18个FER抗病QTL。其中,不同的群体均存在相同的定位区间,与、有重叠区间,与在8.05bin有重叠区间,与在4.06—4.07bins重叠。另外,、、、和分别与之前通过病害评级的方法定位到的、、、和20的定位区间重叠,而且来源于相同的抗源。QTL的定位区间在不同的群体中可以检测到,利用不同的定位方法也可以检测到,说明这些重叠区间可能存在FER的抗性位点。不同群体中和不同方法中的重叠性在一定程度上验证了定位区间的真实性,也说明利用图像分析的方法进行FER抗病QTL定位的可行性。
[1] ULLSTRUP A J. An undescribed ear rot of corn caused by. Phytopathology, 1946, 36: 201-212.
[2] BEZUIDENHOUT H, MAXASAS W F O. Botryosphaeria zeae: the cause of grey ear rot of maize () in South Africa. Phytophylaetica, 1978, 10(1): 21-24.
[3] KUMAR V, SHETTY H S. A new ear and kernel rot of maize caused bypers. ex Fries. Current Science, 1982, 5(12): 620-621.
[4] 张艳, 谭静. 玉米穗粒腐病的研究进展. 现代农业科技, 2014, 21(1): 121-125.
ZHANG Y, TAN J. Research progress on ear rot in maize. Modern Agricultural Science and Technology, 2014, 21(1): 121-122. (in Chinese)
[5] 任金平. 玉米穗腐病研究进展. 吉林农业科学, 1993(3): 39-43.
REN J P. Progress in researching of ear rot of maize. Jilin Agricultural Sciences, 1993(3): 39-43. (in Chinese)
[6] 胡南, 章红. 吉林省玉米穗腐病病原真菌中镰刀菌毒素的研究. 玉米科学, 1997, 5(2): 66-68.
HU N, ZHANG H. A study on production of threeof corn ear rot pathogenic fungi in Jilin province. Journal of Maize Sciences, 1997, 5(2): 66-68. (in Chinese)
[7] WANG J H, NDOYE M, ZHANG J B, LI H P, LIAO Y C. Population structure and genetic diversity of thespecies complex. Toxins, 2011, 3(8): 1020-1037.
[8] 王晓鸣, 石洁, 晋齐鸣, 李晓, 孙世贤. 玉米病虫害田间手册. 北京: 中国农业科学技术出版社, 2010: 55-61.
WANG X M, SHI J, JIN Q M, LI X, SUN S X.. Beijing:China Agricultural Science and Technology Press, 2010: 55-61. (in Chinese)
[9] 徐书法, 陈捷, 高增贵, 邹庆道, 纪明山, 刘海南. 中国玉米茎基腐病和穗腐病研究进展. 植物病理学报, 2006, 36(3): 193-203.
XU S F, CHEN J, GAO Z G, ZOU Q D, JI M S, LIU H N. Maize stalk rot and ear rot in China. Acta Phytopathologica Sinica,2006, 36(3): 193-203. (in Chinese)
[10] 秦子惠, 任旭, 江凯, 武小菲, 杨知还, 王晓鸣. 我国玉米穗腐病致病镰孢种群及禾谷镰孢复合种的鉴定. 植物保护学报, 2014, 41(5): 589-596.
QIN Z H, REN X, JIANG K, WU X F, YANG Z H, WANG X M. Identification ofspecies andspecies complex causing maize ear rot in China. Journal of Plant Protection, 2014, 41(5): 589-596. (in Chinese)
[11] 孙华, 张海剑, 郭宁, 石洁, 陈丹, 马红霞. 黄淮海夏玉米主产区穗腐病病原菌的分离鉴定. 植物保护学报, 2017, 44(5): 796-802.
SUN H, ZHANG H J, GUO N, SHI J, CHEN D, MA H X. Isolation and identification of pathogens causing maize ear rot in Huang-Huai- Hai summer corn region. Journal of Plant Protection, 2017, 44(5): 796-802. (in Chinese)
[12] HAAFSMA A W, LIMAY-RIOS V, TAMBURIC-ILINCIC L. Mycotoxins andspecies associated with maize ear rot in Ontario, Canada. Cereal Research Communications, 2008, 36: 525-527.
[13] 孙华, 张海剑, 马红霞, 石洁, 郭宁, 陈丹, 李坡. 春玉米区穗腐病病原菌组成、分布及禾谷镰孢复合种的鉴定. 物病理学报, 2018, 48(1): 8-15.
SUN H, ZHANG H J, MA H X, SHI J, GUO N, CHEN D, LI P. Composition and distribution of pathogens causing ear rot in spring maize region and identification ofgraminearum species complex. Acta Phytopathologica Sinica, 2018, 48(1): 8-15. (in Chinese)
[14] ATANASOVA V, PONS S, PINSON L, PICOT A, RICHARD F. Chlorogenic acid and maize ear rot resistance: a dynamic study investigatingdevelopment, deoxynivalenol production and phenolic acid accumulation. Molecular plant-microbe interactions, 2012, 25(12): 1605-1616.
[15] 张婷, 孙晓东, 吕国忠. 我国东北地区玉米穗腐镰孢菌的种类及其分离频率. 菌物研究, 2011, 9(1): 9-14.
ZHANG T, SUN X D, Lü G Z.species and its isolation frequency from rot ears of maize in northeast China. Journal of Fungal Research, 2011, 9(1): 9-14. (in Chinese)
[16] 潘惠康, 张兰新. 玉米对穗粒腐病菌的抗病性. 华北农学报, 1987, 2(3): 86-89.
PAN H K, ZHANG L X. Disease resistance of corn to ear and kernel rot. Acta Agriculturae Boreali-Sinica, 1987, 2(3): 86-89. (in Chinese)
[17] 刘春元, 李洪连, 吴建宇, 刘建华. 穗粒腐病菌对玉米幼苗的致病性研究. 河南农业科学, 2005, 11: 58-62.
LIU C Y, LI H L, WU J Y, LIU J H. Studies on pathogenicity of pathogen of ear and seed rot in maize hybrids to maize seedling blight. Journal of Henan Agricultural Sciences, 2005, 11: 58-62. (in Chinese)
[18] 陈广泉. 河西走廊玉米穗粒腐病侵染规律及发病因子研究. 玉米科学, 2006, 14(1): 158-160.
CHEN G Q. Study on infection law and disease factor of corn spike kernel rotten in Hexi corridor. Journal of Maize Sciences, 2006, 14(1): 158-160. (in Chinese)
[19] 刘振库, 贾娇, 苏前富, 孟玲敏, 晋齐鸣. 齐齐哈尔玉米穗腐病病原菌的鉴定和致病性测定. 吉林农业科学, 2014, 39(6): 28-30.
LIU Z K, JIA J, SU Q F, MENG L M, JIN Q M. Identification of pathogen and pathogenicity of maize ear rot in Qiqihaer. Journal of Northeast Agricultural Sciences, 2014, 39(6): 28-30. (in Chinese)
[20] ZHOU D N, WANG X M, CHEN G K, SUN S L, YANG Y, ZHU Z D, DUAN C X. The majorspecies causing maize ear and kernel rot and their toxigenicity in chongqing, China. Toxins, 2018, 10(2): 90-103.
[21] 席靖豪, 赵清爽, 林焕洁, 袁虹霞, 丁胜利, 李洪连. 河南省及周边地区玉米穗腐病病原菌的分离及鉴定. 河南科学, 2018, 36(5): 688-692.
XI J H, ZHAO Q S, LIN H J, YUAN H X, DING S L, LI H L. Isolation and characterization of fungal pathogenic species causing maize ear rot in Henan and beyond provinces. Henan Science, 2018, 36(5): 688-692. (in Chinese)
[22] CHEN J F, SHRESTHA R, DING J Q, ZHENG H J, MU C H, WU J Y, GEORGE M. Genome-wide association study and QTL mapping reveal genomic loci associated withear rot resistance in tropical maize germplasm. Genes Genomes Genetics, 2016, 6(12): 3803-3815.
[23] COAN M M D, SENHORINHO H J C, PINTO R J B, SCAPIM C A, TESSMANN D J, WILLIAMS W P, WARBURTON M L. Genome-wide association study of resistance to ear rot byverticillioides in a tropical field maize and popcorn core collection. Crop Science, 2018, 58(2): 564-578.
[24] YAO L S, LI Y M, MA C Y, TONG L X, DU F L, XU M L. Combined genome-wide association study and transcriptome analysis reveal candidate genes for resistance toear rot in maize. Journal of Integrative Plant Biology, 2020, 62(10): 1535-1551.
[25] GUO Z F, ZOU C, LIU X G, WANG S H, LI W X, JEFFERS D, FAN X M, XU M L, XU Y B. Complex genetic system involved inear rot resistance in maize as revealed by GWAS, bulked sample analysis, and genomic prediction. Plant Disease, 2020, 104(6): 1725-1735.
[26] ZILA C T, SAMAYOA L F, SANTIAGO R, BUTRON A, HOLLAND J B. A genome-wide association study reveals genes associated withear rot resistance in a maize core diversity panel. G3:Genes Genomes Genetics, 2013, 3: 2095-2104.
[27] JU M, ZHOU Z J, MU C, ZHANG X C, GAO J Y, LIANG Y K, CHEN J F, WU Y B, LI X P, WANG S W, WEN J J, YANG L M, WU J Y. Dissecting the genetic architecture ofseed rot resistance in maize by combining QTL mapping and genome-wide association analysis. Scientific Reports, 2017, 7(1): 1109-1115.
[28] PEREA B D, HEFFERS D, GONZALEZ D, KHAIRALLAH M, CORTES M, VELAZQUEZ G, AZPIROZ S, SRINIVASAN G, QTL mapping ofear rot resistance in highland maize, Mexico.Agrociencia, 2001, 35: 181-196.
[29] BUTRON A, SANTIAGO R, CAO A, SAMAYOA L F, MALVAR R A, QTLs for resistance toear rot in a multiparent advanced generation intercross (MAGIC) maize population. Plant Disease, 2019, 103: 897-904.
[30] ROBERTSON L A, JINES M, BALINT P J, KLEINSCHMIDT C E, WHITE D G, PAYNE G A, MARAGOS C M, MOLNAR T L, HOLLAND J B, QTL mapping forear rot and fumonisin contamination resistance in two maize populations. Crop Science, 2006, 46(4): 17434-1743.
[31] LI Z M, DING J Q, WANG R X, CHEN J F, SUN X D, CHEN W, SONG W B, DONG H F, DAI X D, XIA Z L, WU J Y, 2011. A new QTL for resistance toear rot in maize. Journal of Applied Genetics, 2011, 52(4): 403-406.
[32] DING J Q, WANG X M, CHANDER S, YAN J B, LI J S, QTL mapping of resistance toear rot using a RIL population in maize. Molecular Breeding, 2008, 22(3): 395-403.
[33] CHEN J F, DING J Q, LI H M, LI H, LI Z M, SUN X D, LI J J, WANG R X, DAI X D, DONG H F, SONG W B, CHEN W, XIA Z L, WU J Y. Detection and verification of quantitative trait loci for resistance toear rot in maize. Molecular Breeding, 2012, 30(4): 1649-1656.
[34] LIU Y B, HU G H, ZHANG A, LOLADZE A, HU Y X, WANG H, QU J T, ZHANG X C, OLSEN M, VICENTE F S, CROSSA J, LIN F, PRASANNA B M. Genome-wide association study and genomic prediction ofear rot resistance in tropical maize germplasm. The Crop Journal, 2020, doi:10.1016/j.cj.2020.08.008.
[35] 贾玉芳. 玉米穗腐病的发病原因及防治措施. 中国农业信息, 2015(7): 51.
JIA Y F. Causes of corn ear rot and its control measures. China Agricultural Informatics, 2015(7): 51. (in Chinese)
[36] ZENG Z B. Precision mapping of quantitative trait loci. Genetics, 1994, 136(4): 1457-1468.
[37] GARCIA S A, THORNSBERRY J M, TH B E. Structure of linkage disequilibrium in plants. Annual Review of Plant Biology, 2003, 54(1): 357-374.
[38] WEN J, SHEN Y Q, XING Y X, WANG Z Y, HAN S P, LI S J, YANG C M, HAO D Y, ZHANG Y. QTL mapping ofear rot resistance in maize. Plant Disease, 2020, doi: 10.1094/PDIS-02-20- 0411-RE.
[39] 张艳, 张叶, 王梓钰, 闻竞, 韩四平, 郭嘉, 邢跃先. 44份玉米自交系对镰孢穗腐病的抗性鉴定. 植物遗传资源学报, 2019, 20(2): 276-283.
ZHANG Y, ZHANG Y, WANG Z Y, WEN J, HAN S P, GUO J, XING Y X. Evaluation of resistance toear rot in 44 maize inbred lines. Journal of Plant Genetic Resources, 2019, 20(2): 276-283. (in Chinese)
[40] 邹成佳, 崔丽娜, 章振羽, 张小飞, 李荣进, 陈耕, 李晓. 玉米自交系对轮枝镰孢菌穗腐病的抗性评价, 西南农学报, 2017, 30: 1346-1349.
ZOU C J, CUI L N, ZHANG Z Y, ZHANG X F, LI R J, CHEN G, LI X. Evaluation of maize inbred lines for resistance toear rot. Southwest China Journal of Agricultural Sciences, 2017, 30: 1346-1349. (in Chinese)
[41] MASCHIETTO V, COLOMBI C, PIRONA R, PEA G, STROZZI F, MAROCCO A, ROSSINI L, LANUBILE A. QTL mapping and candidate genes for resistance toear rot and fumonisin contamination in maize. BMC Plant Biology, 2017, 17(20): 20.
[42] ALESSANDRA L, VALENTINA M, BORRELLI V M, LORENZO S, LOGRIECO A F, ADRIANO M. Molecular basis of resistance toear rot in maize. Frontiers in Plant Science, 2017, 8: 1774.
[43] JONG G D, PAMPLONA A K A, PINHO R G, BALESTRE M. Genome-wide association analysis of ear rot resistance caused byin maize. Genomics, 2018, 110(5): 291-303.
[44] KING S B, SCOTT G E. Genotypic differences in maize to kernel infection bymoniliforme. Phytopathology, 1981, 71: 1245-1247.
QTL Mapping of Resistance toEar Rot in Maize Based on Image Analysis
WEN Jing, SHEN YanQi, WANG ZiYu, LI ShiJie, MO LanYue, LEI YuHao, ZHANG Yan, HAN SiPing
Institute of Agricultural Biotechnology, Jilin Academy of Agricultural Sciences, Changchun 130033
【】Ear rot in maize is a kind of fungal disease that is prevalent and giving serious threat to maize production worldwide, among which,ear rot (FER) is the most commonly reported in China. The location of FER disease resistance QTL is very important for the genetic improvement of maize ear rot. Locating FER disease-resistant QTL by an image analysis method not only explores a new disease evaluation method for FER in maize, but also validated the FER resistance QTLs located in the early stage through the disease rating method. 【】In this study, three F2populations (namely F2-C, F2-D and F2-J) and their corresponding F2:3families were used for QTL mapping of resistance to FER, which were constructed from a highly FER-susceptible inbred line ZW18 and three highly FER-resistant inbred lines (Cheng 351, Dan 598 and JiV203) respectively. The percentage of diseased spots was obtained by image analysis for the ears of F2:3families, then the FER resistance QTLs were mapped.【】18 FER resistance QTLs were mapped. Among them,,andlocated in 2.02-2.03 bins, 4.06-4.07 bins and 8.06 bin explained the phenotypic variation as high as 21.80%, 25.80% and 27.40%, respectively. Theof the F2-J population and theof the F2-C population and theof the F2-D population all have an overlapping interval, and the explainable phenotypic variation rate reached 16.50%. Thefrom the F2-D population and thefrom the F2-J population have an overlapping interval in 8.05 bin, and thefrom the F2-C population and thefrom the F2-J population have an overlapping interval in the 4.06-4.07 bins of 4thchromosome.In addition, compared with the results of the previous positioning by the disease rating method,,andoverlap with the disease resistance sitelocated by the rating method at 1.06-1.07 bins, andandat 4.06-4.07 bins and the resistance location located by the rating method. The disease siteandoverlap,andcompletely overlap in the 2.04 bin location interval,andare located in the overlap interval of 9.03-9.05 bins in the S2 duplication, and are derived from the same source of resistance.【】The 18 FER resistance sites located in 3 populations, among which the resistance sites located in 1.04-1.07 bins, 4.06-4.07 bins and 8.05 bin can be detected in different populations, located in 2.04 bin and 9.03-9.05 bins can be detected by different detection methods, indicating that FER resistance sites may exist in these intervals. The overlap of QTL positioning intervals in different populations verifies the authenticity of the positioning intervals to a certain extent. The overlapping intervals are located between different methods, indicating that the image analysis method is used to locate FER disease-resistant QTLs with certain accuracy.
maize;; resistance; QTL

10.3864/j.issn.0578-1752.2021.13.003
2020-12-09;
2021-01-18
中央引导地方科技发展资金吉林省重点实验室基础研究专项(202002072JC)、2020年吉林省预算内基本建设资金(2020C019-4)
闻竞,E-mail:jlruby1988@126.com。通信作者韩四平,E-mail:hansp@cjaas.com
(责任编辑 李莉)
猜你喜欢
中华医学图书情报杂志(2022年1期)2022-11-18
中国现代医生(2022年21期)2022-08-22
西南农业学报(2022年5期)2022-06-06
今日农业(2022年4期)2022-06-01
热带亚热带植物学报(2022年2期)2022-04-14
广东农业科学(2021年3期)2021-04-23
天津医科大学学报(2021年1期)2021-01-26
三农资讯半月报(2020年2期)2020-03-09
发明与创新·大科技(2019年5期)2019-07-31
热带农业科技(2019年1期)2019-01-14
