
铁死亡在肺部疾病中的研究进展
2021-06-30 13:50李琦唐以军王梅芳
临床肺科杂志 2021年7期
李琦 唐以军 王梅芳
作者单位:442000 湖北 十堰,湖北医院学院附属十堰市太和医院呼吸与危重症医学科
从细胞到生物体,死亡是所有生命的共同命运。细胞死亡对于细胞和机体正常发育及维持内环境稳态是必不可少的,当细胞死亡调节失调时,会导致各种病理后果。近年来,随着研究的深入,研究者根据细胞死亡时不同的形态学特征提出了坏死性凋亡、自噬性死亡、焦亡和铁死亡等新的死亡类型。其中铁死亡是2012年Dixon等人发现的一种调节性细胞死亡(RCD)形式,在多种疾病中扮演重要角色[1]。目前发现铁死亡在肿瘤、神经退行性疾病、脑损伤、缺血再灌注损伤、动脉粥样硬化、糖尿病、急性肾功能衰竭等疾病中发挥重要作用。近几十年来,由于人为和自然因素造成的环境问题,导致恶性和非恶性呼吸系统疾病的发病率急剧上升,而铁死亡的发现可能为解决呼吸系统疾病提供新思路。
铁死亡概述
一、铁死亡定义及概述
铁是人体中含量最丰富的金属元素之一,是维持人体细胞生长及生命活动所必需的微量元素。它在细胞氧的运输、DNA的生物合成、ATP的合成及电子传递过程中都起着极其重要的作用[2]。此外,各种原因所致的铁过量,特别是二价铁(Fe2+)过量,极大的加速了人类饱和脂肪酸的脂质过氧化[3]。在线粒体氧化磷酸化过程中,细胞在产生ATP的同时产生活性氧(ROS),过量的ROS可导致氧化应激反应,直接或间接损害蛋白质、核酸和脂质等生物分子的合成及代谢过程,导致细胞损伤或者死亡,这种新发现的细胞死亡形式称为铁死亡[1,4]。铁死亡不同于凋亡、坏死等细胞死亡方式,是铁依赖的脂质过氧化和ROS积累为特征的新型细胞死亡形式(见图1)。
铁死亡与凋亡、坏死和自噬在形态学、生物化学和调控机制上均有明显区别。在电子显微镜下铁死亡不会出现类似凋亡过程中发生的染色质凝集、线粒体膜电位消失,坏死过程中发生的质膜破裂,或者自噬过程中出现的双层自噬空泡形成的形态学变化;相反,它主要表现为细胞质膜完整,线粒体变小、膜密度增加、线粒体嵴减少或消失[1]。铁死亡在生物化学上主要表现为细胞脂质过氧化水平增加、ROS增加、细胞内铁增加,谷胱甘肽耗竭,谷胱甘肽过氧化物酶4(GPX4)活性下降等。铁死亡在遗传水平上涉及多种基因,尤以IREB2、RPL8、CS、ATP5G3、TTC35、ACSF2等调节铁代谢基因显著[1]。铁死亡的激活剂包括Erastin、柳氮磺胺吡啶、索拉非尼、青蒿琥酯、顺铂、对乙酰氨基酚等,抑制剂包括去铁胺(DFO)、甲磺酸去铁胺等铁离子螯合剂、Feroptosis-1(Fer-1)、Liproxstatin-1(Lip-1)、维生素E、生育酚等亲脂性抗氧化剂(见图1),坏死、凋亡、自噬等抑制剂均不能抑制铁死亡的发生。
二、铁死亡的调控机制
1 铁代谢
铁是通过Fenton反应催化脂质ROS生成的重要因素,对细胞发生铁死亡起着至关重要的作用。正常组织中铁通过微妙的铁转运系统保持平衡。细胞外的铁,通过转铁蛋白(TF)和转铁蛋白受体(TFR)进入细胞内,进入细胞内的铁主要以铁蛋白的形式储存,细胞内多余的铁由膜铁转运蛋白(FPN)输出细胞,再次参与体内的铁循环,以维持细胞内的铁稳态。Hepcidin由肝细胞产生是全身铁稳态的关键调节因子,FPN是哺乳动物中目前唯一已知的调控细胞内铁输出的蛋白[5]。Hepcidin通过与FPN结合,导致其水解,从而减少FNP介导的细胞内铁向细胞外转移,导致细胞内铁含量增加[6]。增加铁摄取或者减少铁输出都会导致细胞内Fe2+增加,Fe2+与过氧化氢(H2O2)发生Fenton反应,导致体内氧化还原反应失调,引起脂质过氧化反应过度,促进细胞发生铁死亡。Fe2+可以通过TF介导的铁摄取或者铁蛋白自噬而升高[7]。核受体辅激活因子4(NCOA4)可特异性的识别、结合铁蛋白,并介导铁蛋白在溶酶体内由Fe3+转化为Fe2+,释放到胞浆中成为游离铁,该过程称为铁自噬[8]。有研究表明敲除NCOA4可降低细胞对铁死亡的敏感性[9]。铁结合调节蛋白2(IREB2)是编码铁代谢的主要调节因子,沉默IREB2可以显著降低细胞对铁死亡的敏感性[1]。此外热休克蛋白β-1(HSPB1)、CDGSH铁硫域蛋白1(C1SD1)、核因子E2相关因子2(Nrf2),也可以通过直接或间接调节铁代谢,进而调节铁死亡[10-12]。
2 氨基酸与谷胱甘肽代谢
氨基酸代谢与铁死亡调节密切相关。在体内氨基酸需要在特定转运蛋白的帮助下进入细胞内。胱氨酸/谷氨酸反向转运体(cystine/ glutamate antiporter system x c-,system xc-)就是这样的氨基酸转运蛋白体之一,其由二硫键连接的调节亚单位溶质载体家族3成员2(SLC3A2)和催化亚单位溶质载体家族7成员11(SLC7A11)组成,可以将细胞外的胱氨酸与细胞内的谷氨酸以1:1的比例交换,将胱氨酸转运到细胞内并转化为半胱氨酸合成谷胱甘肽[13]。有研究表明细胞外谷氨酸浓度升高可以抑制XC-系统,诱导细胞发生铁死亡。在XC-敲除的小鼠中,细胞外谷氨酸浓度降低,可免受神经毒物的伤害[14]。
在哺乳动物细胞中,谷胱甘肽主要以还原型谷胱甘肽(GSH)和氧化型谷胱甘肽(GSSG)形式存在。谷胱甘肽含量通过合成、利用、代谢和外排的调节方式处于动态平衡[15]。在生理条件下,GSH是主要形式,占98%以上,而GSSG不到1%。GSH参与许多细胞代谢活动,包括ROS的清除,DNA和蛋白质的合成,以及信号转导[16],是哺乳动物对抗氧化应激的关键物质。大部分由谷氨酸、半胱氨酸和甘氨酸在谷氨酸-半胱氨酸连接酶(GCL)和谷胱甘肽合成酶(GSS)的催化下产生[17]。因此,胱氨酸和半胱氨酸的利用率影响GSH的生物合成。铁死亡可以由抑制谷胱甘肽的生物合成或阻断细胞从外环境获取胱氨酸,进而导致GSH耗竭来触发。
3 脂质代谢
脂质代谢也与铁死亡密切相关。与不饱和及单不饱和脂肪酸相比,多不饱和脂肪酸(PUFA)对脂质过氧化更敏感,多不饱和脂肪酸的数量和定位决定了细胞中发生的脂质过氧化程度,向细胞提供PUFA可以提高它们对铁死亡的敏感性[18]。游离PUFA是合成脂质信号介体的底物,然而它们必须酯化成膜磷脂并经历氧化才能传递铁死亡信号。因此,这些PUFA的辅酶衍生物的形成及其与磷脂结合是产生铁死亡信号的必要条件。两种酶,酰基辅酶A合成酶长链家族成员4(ACSL4)和溶血磷脂酰胆碱酰基转移酶3(LPCAT3)参与细胞膜中多不饱和脂肪酸-PE(PUFA-PE)的生物合成和重塑。随着ACSL4的激活,游离的PUFA可以在LPCAT3的帮助下酯化,并结合到膜磷脂中,生成PUFA-PE。因此ACSL4的上调被认为是铁死亡的生物标志物和贡献者。PUFA-PE可以通过促进脂氧合酶(LOXs)介导的酶促反应形成脂质氢过氧化物,细胞内LOXs的耗尽可以防止erastin诱导的铁死亡,这表明LOXs促进细胞发生铁死亡[19]。
4 其他机制
在哺乳动物体内GPX4可以通过两个GSH分子作为电子供体将有毒的脂质过氧化氢还原为无毒的磷脂醇,并产生氧化型谷胱甘肽(GSSG)与脂质过氧化作用对抗[20]。药物抑制或遗传学上GPX4低表达都可以促进ROS的产生,进而诱导细胞发生铁死亡[21]。除GPX4外,抗Nrf2的过度激活可能通过血红素加氧酶1(HO-1)介导的亚铁离子催化ROS代谢来诱导铁死亡[22]。Nrf2也可以通过上调系统X c-来保护细胞避免发生铁死亡[23]。甲羟戊酸途径可以合成辅酶Q10,辅酶Q10在细胞膜上具有抗氧化功能,是一种内源性铁死亡的抑制剂[24]。
铁死亡与呼吸系统疾病
一、铁死亡与COPD
慢性阻塞性肺疾病(COPD)是一种以进行性、不可逆性气流受限为特征的慢性肺部疾病,预计到2020年将成为全球第四大死亡原因[25]。尽管COPD的病因众多,但吸烟仍是COPD最重要的病因。目前有研究表明铁死亡与吸烟及COPD的发生发展有密切联系,为探究COPD新的防治方法提供了方向。
Park等用整支香烟烟雾冷凝物(WCSC)处理人支气管上皮细胞BEAS-2B细胞,发现WCSC处理的细胞线粒体浓缩,体积减少呈现出铁死亡的形态特征。对WCSC处理后的细胞总RNA进行微阵列分析发现铁结合核蛋白(PIR)的基因表达上调最多,NQO1、Hmox1等抗氧化相关基因的表达明显增强,对差异表达基因进行富集分析发现WCSC可激活铁死亡关信号通路,对上调基因进行信号通路分析发现铁的吸收/释放和氧化应激途径与之密切相关[26]。Yoshida等发现香烟烟雾(CS)诱导人支气管上皮细胞(HBECs)24 h后细胞内呈现明显的脂质过氧化,电镜下细胞呈现铁死亡的形态学特征,使用铁螯合剂DFO、特异性的铁死亡抑制剂Fer-1和Lip-1分别预处理细胞后可以明显改善CS诱导的脂质过氧化、铁离子增加和细胞死亡,而坏死抑制剂necrostatin-1和凋亡抑制剂zVAD-FMK对CS所致的细胞死亡改善不明显。敲除GPX4可显著增强CSE诱导的HBECs和A549细胞内的脂质过氧化和细胞死亡,且细胞内线粒体膜密度增加更为明显,这些研究表明在体外实验中,铁死亡是CS诱导支气管上皮细胞死亡的主要原因。在体内实验中,暴露于CS的GPX4基因敲除鼠较野生型小鼠支气管上皮细胞游离铁增加,小鼠气道和肺组织均浆中游离铁水平明显增高,且小鼠肺组织中气道上皮细胞、肺泡上皮细胞死亡明显增加,灌洗液中炎性细胞及促炎因子表达明显增加,而GPX4基因过表达的小鼠,以上特征均明显减弱。在人体中COPD患者的HBECs中GPX4明显低于从不吸烟者和吸烟的非COPD患者,且GPx4的表达水平与FEV1/FVC的百分率呈正相关,电镜下COPD患者的气道上皮细胞表现出明显的铁死亡形态学特征,而不吸烟者的气道上皮细胞不会表现出这种变化[27]。Sampilvanjil 等的研究进一步证明铁死亡在香烟提取物(CSE)诱导的平滑肌细胞死亡中发挥重要作用[28]。这些研究表明铁死亡在COPD发生发展中可能扮演着至关重要的作用,铁死亡抑制剂可能是治疗及阻止COPD进展的一个新疗法。
二、铁死亡与肺纤维化
肺纤维化是一种慢性进行性的间质性肺病,金属粉尘、灰尘、射线等职业环境,病毒感染、胃食管反流等病理因素都可以导致肺纤维化的发生,还有一部分病因不明的称为特发性肺纤维化(IPF)。肺纤维化最显著的病理特征是纤维化病灶的形成,在肺内成纤维细胞分化为肌成纤维细胞,肌成纤维细胞导致大量细胞外基质沉积,胶原积聚,肺泡结构破坏最终导致正常肺组织结构破坏[29]。研究表明,IPF患者肺中的总铁水平、铁相关氧自由基和含铁巨噬细胞较对照组均有所增加[30]。肺内过量铁的积累已被证明与IPF患者肺部的血管异常、肺动脉高压相关,这就为肺纤维化过程中发生铁死亡提供了条件。
Gong Yue等的研究表明TGF-β1处理的HFL1细胞a-SMA、COL I表达升高,呈现纤维化的特征,同时电镜下该细胞线粒体体积缩小、线粒体嵴减小,GPX4降低,呈现铁死亡的特征,Fer-1可以抑制TGF-β1诱导的ROS、MDA、a-SMA、COL I升高和GPX4降低,说明Fer-1可以通过抑制脂质过氧化和促进GPX4的表达,来抑制铁死亡进而抑制成纤维细胞向肌成纤维细胞的分化[31]。Li Xuan等利用C57BL/6小鼠构建放射性肺纤维化模型,与对照组相比实验组小鼠血清炎性细胞因子水平明显增加,肺组织中HYP、胶原沉积明显增加,肺组织切片呈现明显的纤维化特征,小鼠肺组织中ROS明显增加,GPX4降低,而铁死亡抑制剂Lip-1可抑制实验组小鼠肺组织胶原沉积,降低HYP、ROS含量,减轻血清炎性因子释放,增加肺组织GPX4水平[32]。Wang Yan等研究发现用PM2.5诱导内皮细胞,导致内皮细胞内铁含量增加、脂质过氧化和氧化还原平衡失调,进而导致细胞发生铁死亡与分泌炎症因子。而Fer-1和铁螯合剂甲磺酸去铁胺减少内皮细胞死亡,降低炎症因子分泌[33]。故铁死亡在肺纤维化的发生发展过程中扮演了重要作用,为治疗肺纤维化提供了一个新思路。
三、急性肺损伤
急性肺损伤(ALI)是一种由各种肺内肺外因素所致的肺部炎症细胞浸润,导致肺组织间质水肿、肺泡-毛细血管屏障功能损害和肺泡上皮损伤,以气体交换障碍为特征的致命疾病。目前已有研究表明铁死亡在ALI的发生发展过程中发挥重要作用。
周航等研究发现在油酸(OA)诱导ALI小鼠模型中,较正常对照组OA组电镜下肺泡II型上皮细胞呈现出线粒体皱缩、线粒体膜破裂的典型铁死亡形态学特征,肺组织中PTGS2mRNA、丙二醛、铁离子含量增加,GSH、GPX4表达降低[34]。Liu Pengfei等通过脂多糖(LPS)诱导建立体内、体外ALI模型,LPS诱导的人支气管上皮细胞中MDA、4-HNE、铁离子、Hepcidin表达增加,SLC7A11、GPX4表达下降,表明LPS诱导人支气管上皮细胞促进其发生铁死亡,而Fer-1可以减少LPS诱导的细胞死亡。在LPS诱导的小鼠模型中,肺组织MDA、4-HNE、铁离子、Hepcidin、PTGS2表达增加,GPX4、SCL7A11表达降低,而Fer-1可以减轻LPS诱导的小鼠肺部炎症反应[35]。邵强等研究发现,在LPS诱导的人支气管上皮细胞中,铁死亡激活剂Erastin能够放大LPS诱导的炎症介质释放[36]。进一步研究表明在ALI中nrf2可以通过促进HO-1和SLC7A11的表达,抑制铁死亡,从而对ALI起到保护作用[37],为治疗ALI提供了新方向。
四、铁死亡与结核
肺结核是由结核分枝杆菌感染所致的一种传染性疾病。从史前以来,结核病就一直威胁着人类健康,至今结核仍是单一感染源造成死亡的主要原因。由于缺乏有效的成人肺结核疫苗,及多耐药结核的发病率逐年增高,研发新的抗结核药物意义重大。
已有大量研究表明,结核与血清铁密切相关。据报道,体内铁水平的增加与患活动性肺结核病风险和结核感染小鼠体内细菌负荷增加相关[38]。相反,影响铁利用率的铁螯合剂和宿主或细菌基因的突变通常会限制结核病原体的生长。此外,结核分枝杆菌感染会诱导血红素加氧酶-1其能将血红素降解为游离铁[39]。Amaral等的研究发现在体外结核分枝杆菌感染小鼠巨噬细胞会促进小鼠巨噬细胞内游离铁、线粒体超氧化物和脂质过氧化增加,GSH和Gpx4水平降低,这些都是铁死亡的重要特征。Amaral等人还发现在低感染复数(MOI=1)时,在无铁培养皿中,结核分枝杆菌感染巨噬细胞不会发生细胞死亡,而在高铁培养皿中细胞24小时死亡明显增加,且伴有线粒体超氧化物和脂质过氧化物的升高;而使用铁螯合剂吡哆醛异烟酰腙(PIH)和Fer-1可以减少结核分枝杆菌感染的巨噬细胞内脂质过氧化从而减少细胞死亡。在结核分枝杆菌感染的人单核细胞来源的巨噬细胞中,Fer-1也可以抑制细胞死亡。在结核分枝杆菌感染的小鼠模型中,感染小鼠肺组织中GPX4mRNA的表达显著低于未感染的动物,感染结核分枝杆菌的小鼠在肺组织匀浆中显示出高水平的脂质过氧化,接受Fer-1治疗的感染小鼠中,Fer-1不仅能减少感染小鼠的肺坏死,而且还能显著地降低感染小鼠肺和脾脏的细菌数量[40]。以上的证据表明,铁死亡与结核分枝杆菌感染的病理过程密切相关,抑制铁死亡可以抑制结核感染及炎症反应。
五、铁死亡与肺癌
肺癌是全球最常见和最致命的恶性肿瘤之一,严重危害人类健康。目前肺癌的治疗手段包括手术切除、放疗、化疗、免疫治疗、分子靶向治疗等,但大部分患者在治疗过程中会出现耐药和复发,导致病情恶化,因此研究肺癌的发生发展及耐药机制十分重要。
已有研究报道,铁代谢失调与肺癌的发生发展密切相关。在肺癌患者的血清、支气管肺泡灌洗液(BAL)和呼气冷凝液中发现铁蛋白水平升高[41]。TFR1在88%的非小细胞肺癌患者中升高,而EGFR通过促进TFR1的高表达调节细胞铁稳态,增加细胞内铁的输入,促进肺癌的发展[42]。研究表明在肺癌组织中GPX4高表达,提示预后不良[43]。近期Alvarez 等研究表明在肺癌细胞系和组织中铁硫簇生物合成酶(NSF1)表达增高,且在高氧环境下NSF1明显缓解了肺癌细胞发生铁死亡,抑制NSF1能诱导肿瘤细胞摄取大量铁并释放细胞内储存铁,与抑制半胱氨酸转运协同作用,促进肿瘤细胞发生铁死亡[44]。Huang Chaoli等发现铁死亡激活剂Erastin可以通过促进p53表达上调,细胞内ROS增加,抑制SLC7A11表达,抑制肺癌细胞增殖[45]。Chen Peng等的研究进一步表明毛兰素(Erianin)处理肺癌细胞后,细胞发生ROS增加、脂质过氧化和GSH耗竭等铁死亡的特征,Fer-1、Lip-1可抑制Erianin诱导的细胞死亡和抑制细胞迁移,而坏死抑制剂、凋亡抑制剂和自噬抑制剂不能抑制Erianin的作用。进一步的研究表明Erianin通过激活Ca2+/CaM信号通路激活铁死亡[46]。Gai Chengcheng等的研究表明Erastin和APAP协同作用,通过调节核因子红系2相关因子2(Nrf2)的核转位促进NSCLC细胞发生铁死亡[47]。Pan Xiaofen等的研究表明Erastin可以通过激活铁死亡增加A549和H460对射线的的敏感性[48]。Li Yu等的研究表明Erastin和sorafenib不仅可以通过联合顺铂诱导肺癌细胞发生铁死亡,而且可以通过抑制nrf2/xCT通路诱导顺铂耐药的肺癌细胞发生铁死亡[49]。上述研究均表明铁死亡在肺癌发生发展及耐药机制方面发挥重要作用,铁死亡有望成为治疗肺癌的新方向。
结束语
越来越多的证据表明,铁死亡与COPD、肺结核、肺癌等多种肺部疾病发生发展密切相关(见表1)。但铁死亡与肺部疾病的研究尚处于早期阶段,大部分研究仅揭示了呼吸系统疾病中存在铁死亡现象,但其具体机制尚不明确。有研究显示黄芩素、槲皮素、青蒿素类衍生物、异甘草酸镁等传统中药组分在COPD、哮喘、肺纤维化、急性肺损伤、肺癌等呼吸系统疾病中均能发挥重要保护作用,而黄芩素、槲皮素等传统中药成分可以抑制细胞发生铁死亡,青蒿素类衍生物、异甘草酸镁等中药成分可以激活铁死亡,这些药物对铁死亡的诱导差异,为我们进一步探究铁死亡诱导剂和抑制剂在呼吸系统疾病的临床应用提供方向。

表1 铁死亡和肺部疾病
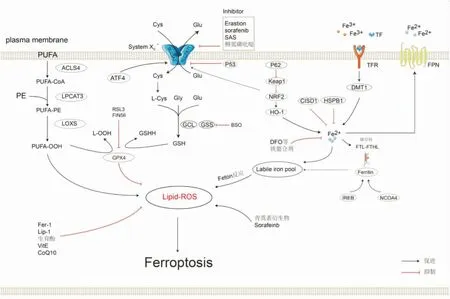
图1 铁死亡分子机制
总之,作为一种新型的细胞死亡形式,铁死亡与肺部疾病的发生发展密切相关,进一步探究铁死亡与疾病发生发展的具体机制和铁死亡在同一疾病不同阶段的干预措施,有助于我们加深对肺部疾病的认识,以期为肺部疾病提供更精准的治疗。
猜你喜欢
中老年保健(2022年2期)2022-11-25
长春中医药大学学报(2022年5期)2022-05-24
昆明医科大学学报(2022年4期)2022-05-23
家庭医学(2022年5期)2022-04-27
现代养生·上半月(2021年10期)2021-09-24
分析化学(2018年4期)2018-11-02
食品界(2018年8期)2018-09-03
现代养生·下半月(2018年8期)2018-03-12
宁波职业技术学院学报(2017年5期)2017-04-09
饮食科学(2016年12期)2017-01-07
