
高效液相色谱法测定液态乳中乳果糖方法的优化
2021-06-22 01:09李盛楠张敏婕胡浩鑫冯沛彦韦晓群
食品工业科技 2021年1期
李盛楠,张敏婕,胡浩鑫,冯沛彦,曾 静,韦晓群,3,*
(1.华南农业大学食品学院,广东广州210095;2.温氏食品集团股份有限公司,广东新兴527400;3.广州广电计量检测股份有限公司,广东广州510656)
非天然存在的乳果糖(Lactulose)由一分子半乳糖和一分子果糖组成,是生牛乳中乳糖经热处理后碱基异构化而形成的双糖产物[1]。生牛乳经不同工艺热处理后的液态乳中乳果糖含量差异明显[2]。因此,可以通过乳果糖含量评价液态乳在热加工处理中的受热强度从而监测产品在热加工过程中的质量变化[3-5]。
随着检测仪器与方法的不断改进,高效液相色谱法(HPLC)以其简便的操作方法和较高分辨率与精密度,逐渐代替酶解比色法成为乳果糖含量检测的主要方法[6-9]。乳果糖与乳糖互为同分异构体[10]且经热处理后在液态乳中含量相差较大,通常乳糖浓度高于乳果糖浓度三个数量级[11],因而导致两者在色谱柱上分离效果不佳,进而影响乳果糖准确定量[12-13]。除此之外,受色谱柱上功能基团化学稳定的影响,采用高效液相色谱法对进行乳果糖定量时可能会产生误差[14],目前用于乳果糖高效液相检测的色谱柱主要以糖基柱[15-16]和氨基柱[17-19]为主,虽然它们能满足液态乳中乳果糖的检测分离需求,但从长期检测出发,两种色谱柱上的功能基团较易损耗,进而影响定量结果的准确。Manzi等[20]采用两根氨基柱对乳果糖进行检测时发现氨基柱上的氨丙基硅烷极性基团极易受盐离子干扰,并与待测糖形成薛夫碱(Schiff),导致定量结果偏低,同时王桂云等[21]利用氨基柱测定几种还原糖时发现氨基柱的键合官能团氨丙基易水解,柱流失严重色谱柱寿命短。Silveira等[15]应用糖分析柱对乳果糖进行检测,发现糖基柱中的分离因子磺酸盐自由基(R-SO-3)极易与Ca2+产生电荷效应,导致柱效降低使待测物各组分保留时间漂移影响定量效果。
改性酰胺柱[22]作为一种化学性质的色谱柱,其中酰胺基可与不同糖醇或者糖醛基形成氢键作用性质稳定,且难以与糖基键合形成希夫碱(Schiff),避免了样品中乳果糖浓度损失,柱流失减少,使用寿命增长,理论上可作为一种乳果糖日常分析检测的分离柱,但考虑到在检测乳果糖时其他二糖也可能与酰胺柱上的活性基团键合从而对乳果糖色谱峰产生干扰因而目前在乳果糖的高效液相色谱分离定量中未见良好应用。
本文拟使用化学稳定的改性酰胺基色谱柱,通过改变进样条件使液态乳中高含量乳糖与低含量乳果糖分离满足定量要求,并且通过探究不同单糖及双糖在改性酰胺基色谱柱上的保留时间规律,寻找对乳果糖色谱分离产生干扰的二糖并提出解决方法,从而建立起一种基于高效液相色谱-蒸发光散射检测器(HPLC-ELSD)更加经济简便、定量准确的日常乳果糖定量方法。
1 材料与方法
1.1 材料与仪器
乳果糖(C12H22O11,CAS No.:4618-18-2)标准品 纯度≥99%,北京索莱宝科技有限公司;葡萄糖淀粉酶 活度2000 U/mL,中国上海ANPEL实验技术有限公司;乙腈(色谱级)美国Thermo Fisher公司;甲醇(MeOH)、乙酸(HAC)分析纯,广州化学试剂厂;超纯水 离子交换系统制备,电阻率>18 mΩ,中国广州实验室;27种牛奶实际样本 广州永旺百货超市。
高效液相色谱仪(岛津Nexera HPLC LC-30A配蒸发光散射检测器) 日本岛津公司;Acchrom Xamide色谱柱(5μm,4.6 mm×250 mm)日本岛津公司;5804R高速离心机 德国Eppendorf公司;R2010旋转蒸发仪 上海霓玥仪器有限公司;HC1204分析天平(精度0.1 mg)上海花潮实业有限公司;W-201B恒温水浴锅 江苏金怡仪器科技有限公司。
1.2 实验方法
1.2.1 标准溶液的配制 准确称取2.5 g乳果糖标准品溶于水中,用水稀释至25 mL,制成100000 mg/L的乳果糖储备液,2~4℃保存,有效期为3个月。1 mL的100000 mg/L乳果糖储备液转移至样品瓶,用缓冲溶液稀释至10 mL,制成10000 mg/L乳果糖标准溶液,2~4℃保存,有效期为3个月。
1.2.2 样品前处理 精确量取24 mL样品(牛奶)于50 mL离心管,加入1 mL乙酸,振摇2 min,以10000 r/min离心,定量滤纸过滤,所得滤液采用0.22μm滤膜过滤后取1 mL于进样小瓶,上机分析。若检测样品经色谱分离发现乳果糖色谱峰被麦芽糖色谱峰所干扰,考虑在样品前处理离心后加入0.2 mL葡萄糖淀粉酶,45℃酶解3 h,酶解产物经0.22μm滤膜过滤,取1 mL于进样小瓶,上机分析;若样品中乳果糖的含量较低,则在离心后70℃旋转蒸发浓缩10倍,后过膜分析。
1.2.3 液相色谱条件 流动相A相:水-B相:乙腈;洗脱程序:17%A(0~25 min),80%A(25.1~30 min),17%A(30.1~40 min)梯度洗脱40 min;色谱柱:Acchrom X-amide(5μm,250 mm×4.6 mm);柱温:25℃;平衡时间:12 min;流速:1.0 mL/min;进样量:3μL;检测器:蒸发光散射检测器(ELSD)。
1.3 数据处理
实验数据统计分析采用SPSS 17.0,色谱采集及处理采用岛津液相色谱仪配套的LC Solution色谱处理系统。
2 结果与分析
2.1 乳果糖与乳糖的分离
当乳糖浓度远高于乳果糖浓度3个数量级时以常规体积进样,二者分离效果不佳[23-24]。杨朝辉[25]研究表明当样品浓度一定时进样体积会对色谱峰宽以及分离度产生影响,因而本实验改变进样量(1、3、4、6、8、10、15μL)试图改善乳果糖和乳糖的分离效果。色谱分离的有效性由分辨率表示,即两个相邻峰的保留时间差与平均峰宽之比。R越大,两个相邻成分的分离效果越好。通常,当R<1时,两个峰之间有一些重叠;当R=1.0时,称为4σ分离,两个峰大部分分离,分离度可达到98%;当R=1.5时,称为6σ分离,分离度可以达到99.7%。R=1.5通常用作两个相邻峰已完全分离的标记[26]。结果表明,当进样量从1μL增至15μL时,分离度从2.2降至0.9(图1),进样体积越小,乳果糖与乳糖分离度越高。大体积进样会造成色谱柱超载,柱效降低,导致峰宽增加分离度降低。因此,降低进样体积可将液态乳中乳糖与乳果糖的有效分离。

图1 进样量对分离度的影响Fig.1 Effect of injection volume on resolution
本实验最终以3μL色谱进样分析,此时乳果糖与果糖分离度R=1.6。两个色谱峰被完全分开(图2)满足定量检测峰面积积分要求。
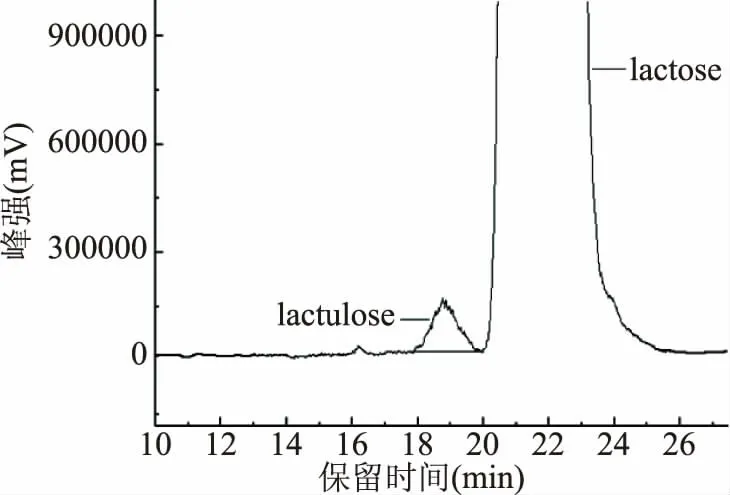
图2 乳果糖与乳糖的色谱分离图Fig.2 Chromatographic separation of lactulose and lactose
2.2 其他二糖分离干扰的排除
黄晓林等[27-28]研究发现常规纯牛奶中只含乳糖和乳果糖,但部分液态乳中会添加蔗糖、麦芽糖等双糖以增加风味,其中部分双糖因会与色谱柱上的酰胺基键合从而干扰乳果糖在色谱柱上的分离。本实验将通过半乳糖、葡萄糖、果糖等单糖及其组合双糖的保留时间规律探究干扰乳果糖分离的双糖。结果显示(图3),单糖的保留时间顺序依次为果糖、葡萄糖和半乳糖,而其组合双糖的保留时间顺序依次为蔗糖(果糖-葡萄糖)、乳果糖(果糖-半乳糖)和乳糖(葡萄糖-半乳糖)。表明不同浓度流动相下双糖的保留时间顺序与其单糖分子的保留时间顺序相同,据此推测乳制品中所含的麦芽糖(葡萄糖-葡萄糖)的保留时间则可能与乳果糖(果糖-半乳糖)的色谱保留时间相同或者接近从而干扰乳果糖在色谱柱上的分离测定。为进一步验证以上推论,使用改性酰胺基色谱柱(5μm,4.6 mm×250 mm),并以乙腈-水(83%~17%)为流动相,1 mL/min流速洗脱30 min。结果显示(图4),乳果糖(Rt=18.9 min)与蔗糖(Rt=16.6 min)、乳糖(Rt=21.0 min,22.2 min)很好地被明显地分离,麦芽糖(Rt=19.7 min,21.0 min)中α-麦芽糖色谱保留时间与乳果糖(Rt=18.9 min)接近,色谱图显示两者未见分离,此时R=0.7,符合上述对于麦芽糖在改性酰胺基色谱柱上可能会对乳果糖的分离产生干扰的推测。
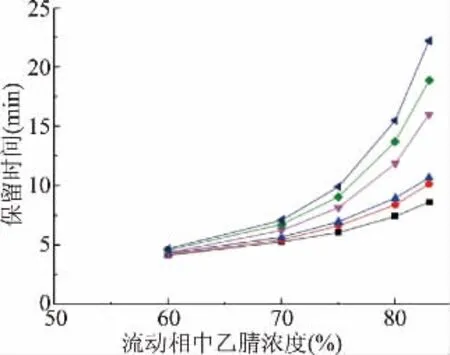
图3 不同流动相下不同糖的的保留时间变化Fig.3 Changes of the retention time of different sugars at various mobile phase

图4 相同条件下麦芽糖与乳果糖保留时间对照Fig.4 Comparison of retention time of maltose and lactulose under the same chromatographic condition
为进一步验证麦芽糖在改性酰胺基色谱柱上的干扰,应用ChemDraw 3D软件通过能量最小化计算三个酰胺基与麦芽糖(图5a)和乳果糖(图5b)形成氢键的类型与数量。计算结果显示两种糖都可以与色谱柱上的三个酰胺基形成4个NH-OH氢键,2个C=O-OH氢键,证明麦芽糖(Rt=19.7 min)和乳果糖(Rt=18.9 min)在改性酰胺基色谱柱上保留时间相近,当麦芽糖和乳果糖同时存在时几乎不可能实现基线分离。因此,若经过检测发现样品中存在麦芽糖时可考虑在样品前处理中加入葡萄糖淀粉酶[29]试图将麦芽糖水解为葡萄糖,以消除α-麦芽糖对乳果糖的色谱峰干扰。
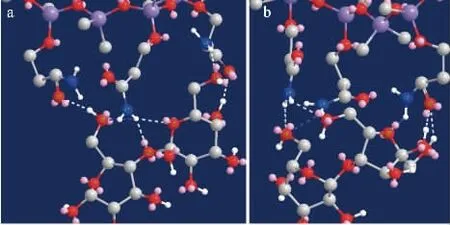
图5 3D建模Fig.5 3D modeling
2.3 方法学验证
2.3.1 标准曲线、检出限(LOD)和定量限(LOQ)将乳果糖标准液浓度依次稀释为50、100、250、500、1000、2500 mg/L,并换算为质量分数,以乳果糖质量分数为横坐标(X,mg/kg),对应峰面积为纵坐标以建立标准曲线(Y),得线性方程Y=1849.1X-52528,该线性在50~2500 mg/kg范围内线性关系良好,R2=0.9997。
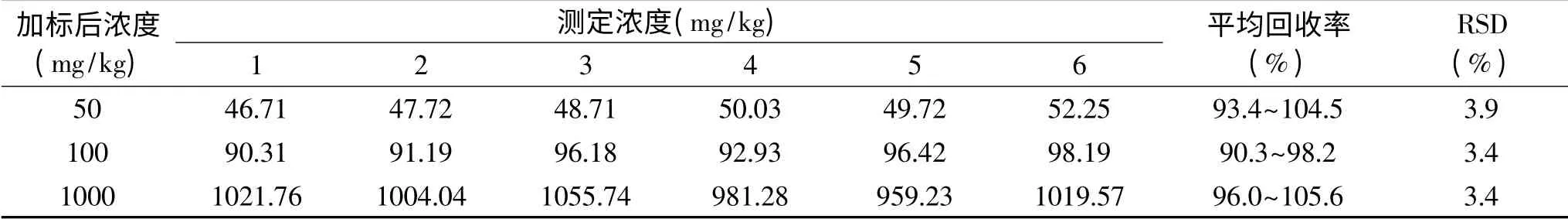
表1 三个水平下乳果糖浓度的回收率和精密度Table 1 Recoveries and RSDs of lactulose concentration at three levels
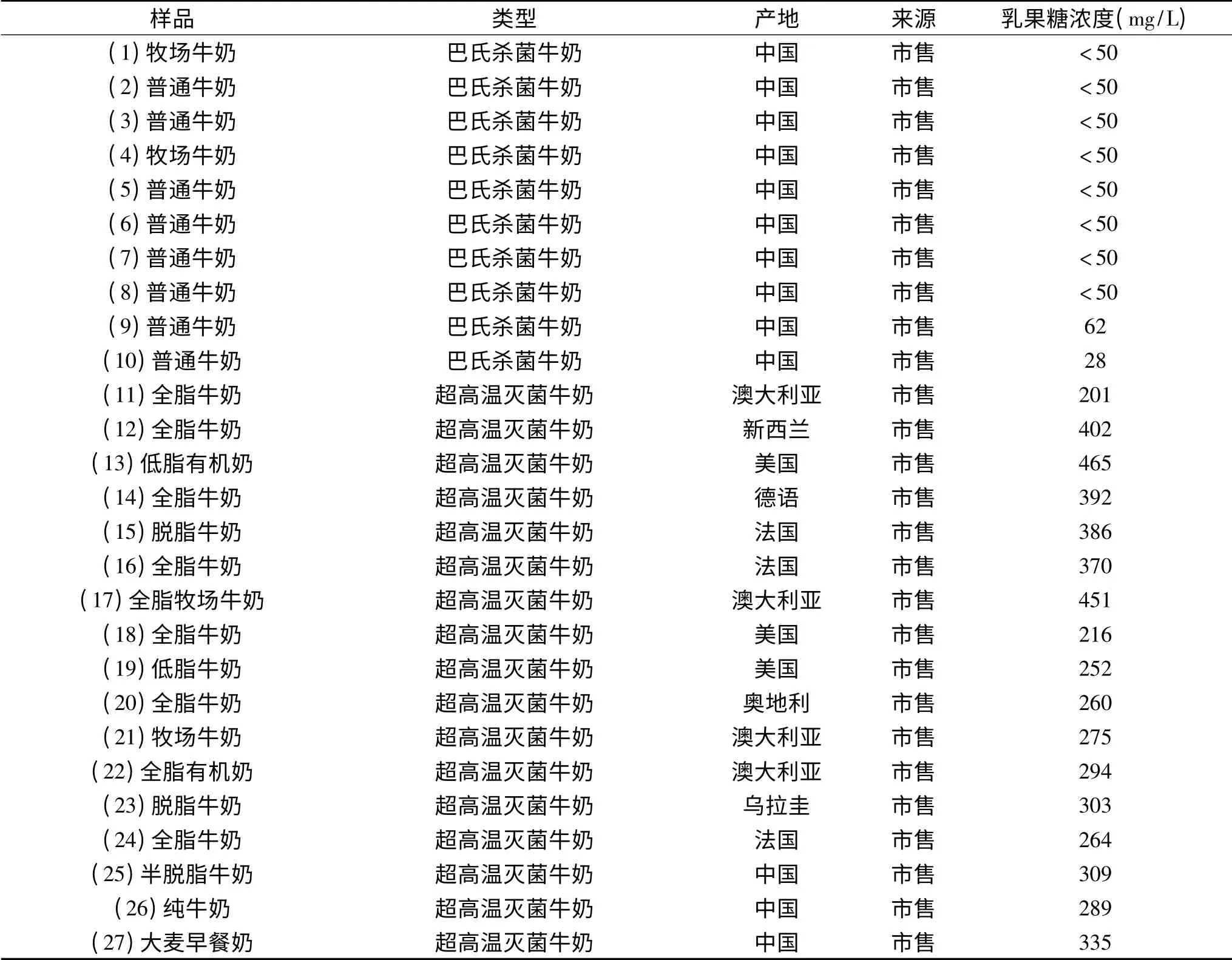
表2 市售牛奶中乳果糖的测定结果Table 2 Results of determination of lactulose in milk sold on the market
逐步稀释乳果糖标准溶液浓度当乳果糖峰高为基线噪音高3倍和10倍时,检测限与定量限分别为15和50 mg/kg。
2.3.2 准确度与精密度 本方法通过在液体中加标方法测定回收率的方法来验证方法的正确度。方法选择1倍定量限、20倍定量限、200倍定量限三个水平进行回收率试验,即样品总添加浓度为50、100和1000 mg/kg,覆盖NY/T939-2016中100~600 mg/kg的范围。每个加标水平测定6次。乳果糖的加标回收率在90.3%~105.6%之间,RSD低于5%,在3.4%~3.9%之间变化(表1),表明该方法准确度和精密度良好,符合分析要求。
2.4 样品检测
根据田瑶[13]对液态乳中乳果糖含量研究表明,高温巴氏杀菌乳中的乳果糖的浓度范围在6.7~80 mg/L,UHT灭菌乳中乳果糖质量浓度为50~1065.1 mg/L,直接UHT灭菌乳中乳果糖质量浓为50~850 mg/L,间接UHT灭菌乳中乳果糖质量浓度为190~1065.1 mg/L,因而采用本方法对10种高温巴氏灭菌牛奶样品和17种UHT牛奶样品进行了分析,结果显示,所有检测结果均在可控合理的范围内。
3 结论
本实验采用化学稳定的改性酰胺基色谱柱完善了液态乳中乳果糖含量的高效液相色谱(HPLC)分析方法。通过探究不同进样体积对色谱的分离效果以及不同流动相比例下单糖的色谱出峰规律首先发现降低进样体积有利于提高乳果糖和乳糖的色谱分离,其次发现α-麦芽糖会对乳果糖的色谱峰产生干扰,因而考虑在样品前处理时加入葡萄糖淀粉酶排除α-麦芽糖的色谱干扰。最终经优化后的方法经方法学验证,乳果糖在50~2500 mg/kg的范围内线性关系良好,R2=0.9997,乳果糖检出限与定量限分别为定量限为15和50 mg/kg加标回收率在90.3%~105.6%之间,相对标准偏差在3.0%~3.9%。运用该方法对市场中的27种实际乳样进行分析结果显示,乳果糖的检测含量均在合理范围内。本方法为液态乳中乳果糖的日常检测提供了一种更加简便经济,灵敏准确的途径,有利于业界以对液态乳加工质量准确评价。
猜你喜欢
保健与生活(2022年13期)2022-07-06
江苏卫生保健(2022年5期)2022-05-24
当代水产(2020年3期)2020-06-15
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
作文与考试·小学高年级版(2017年24期)2017-12-14
老友(2016年3期)2016-05-26
恋爱婚姻家庭·养生版(2016年5期)2016-05-06
饮食科学(2015年4期)2015-11-28
火炸药学报(2014年3期)2014-03-20
