
基于不同靶点及治疗策略的嵌合抗原受体T 细胞疗法在实体瘤中应用的研究进展
2021-06-18 01:45:58姜凤婷郑眉综述周旭晏子厚审校
中国生物制品学杂志 2021年6期
姜凤婷,郑眉 综述,周旭,晏子厚 审校
上海生物制品研究所有限责任公司,上海200050
根据世界卫生组织 / 国际癌症研究署(World Health Organization / The International Agency for Research on Cancer,WHO / IARC)发表的最新全球癌症统计数据显示,2018 年全球约有1 807 万新增癌症病例,955 万死亡病例[1]。我国新发癌症428.5万例,死亡病例286.5 万例,其中肺癌、结直肠癌、胃癌、肝癌及乳腺癌等常见实体瘤的发病率占我国新发癌症病例数的58.6%[1]。肺癌仍居全球癌症发病率和死亡率之首;乳腺癌是威胁女性健康的重要病因[2];胰腺癌是预后最差的恶性肿瘤之一,5年生存率仅9%[3]。诸多肿瘤致病因素复杂多样,治疗方法不尽相同,临床治疗仍以手术切除为主,结合放、化疗及其他治疗方法(如癌症疫苗、免疫检查点抑制剂等),但目前并未找到攻克癌症的最佳手段。近年来,以嵌合抗原受体T 细胞(chimeric antigen receptor T cells,CAR-T)为代表的肿瘤细胞免疫疗法为肿瘤治疗带来新的希望。
1 CAR-T 的简介
CAR-T 作为肿瘤免疫治疗的新兴手段之一,已成为近年来肿瘤治疗研究领域的研究焦点和热点。CAR-T 是利用基因工程技术,将患者自身的T 细胞分离体外,并在其表面表达一个能特异性识别肿瘤表面抗原的单链抗体(single chain antibody fragment,ScFv)区域,实现精准靶向杀伤癌细胞[4]。基因编辑后的T 细胞表达一个CAR 分子,主要由胞外ScFv 区、跨膜区及胞内信号传导区3 部分组成[5]。经不断优化,产生了5 代CAR-T:1 代CAR-T 仅有单一的胞内信号分子CD3ζ,T 细胞无法有效增殖,影响其杀伤肿瘤持久性,疗效有限;2 代CAR-T 引入一种共刺激信号分子,如CD28、4-1BB,CAR-T 细胞抗肿瘤活性和持久性得到明显提高;为进一步增强CAR-T 细胞杀伤能力,3 代CAR-T 又增加了一个共刺激分子,即同时添加CD28 和4-1BB;4 代CAR-T 通过联合趋化因子、细胞因子等改善肿瘤免疫微环境,进一步增强了CAR-T 细胞的活性及穿透肿瘤细胞的能力,实现更加强大的杀伤作用;5 代CAR-T 又称为“通用型CART”,通过敲除内源性T 细胞受体(T cell receptor,TCR)和人类白细胞抗原I 类分子(human leukocyte antigen-I,HLA-I)降低异体移植的免疫排斥风险。
2 CAR-T 在实体瘤中的应用
CAR-T 应用实体瘤的疗效低于血液肿瘤。与B细胞表面的膜抗原CD19 不同,几乎所有实体瘤细胞表面高表达的抗原在多种正常组织中均有不同程度的表达,目前仍未发现在肿瘤细胞表面特异性表达的抗原,导致CAR-T 细胞在用于实体瘤时会产生一系列问题。目前认为,限制CAR-T 细胞杀伤实体瘤活性的因素主要包括:肿瘤细胞表面缺乏特异性抗原、肿瘤抗原逃逸现象、脱靶效应、肿瘤微环境抑制、CAR-T 细胞难以归巢导致其无法到达肿瘤部位发挥作用(如实体瘤细胞分泌趋化因子CXCL12 和CXCL5,抑制CAR-T 细胞抵达肿瘤部位)、细胞因子风暴、神经毒性症状、肿瘤溶解综合症等[6-7]。
由于不同实体肿瘤组织来源各异,结构复杂,发现针对某种实体瘤细胞表面高表达的肿瘤特异性抗原(tumor specific antigen,TSA)成为难点。因此,寻找更多的肿瘤相关抗原(tumor-associated antigen,TAA)成为治疗取得成功的重要突破口和亟待解决的问题。近年,研究人员也发现了一些针对实体瘤的潜在靶点。
2.1 间皮素(mesothelin,MSLN) MSLN 是由糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)锚定的糖蛋白,相对分子质量约40 000。在正常组织细胞中不表达或微量表达于间皮细胞。几乎100%的胰腺癌均为MSLN 阳性,其他肿瘤如肝外胆管癌(95%)、子宫内膜癌(89%)、三阴性乳腺癌(66%)、食道癌(46%)、结直肠癌(30%)和宫颈癌(25%)等均有不同程度表达[8]。异常MSLN 高表达使肿瘤进一步恶化,发生途径主要有两种:一是通过GPI 激活胞内信号通路,并持续活化 NF-κB、MAPK 和 PI3-激酶信号通路,促进肿瘤细胞增殖,并增强其抗凋亡能力;二是 MSLN 与受体 CA125 / MUC16 发生高亲和力结合,促进细胞间异型黏附,导致肿瘤细胞发生扩散及转移[9]。因此,根据MSLN 相对高表达,可作为CAR-T 细胞治疗的特异性靶点。
2.2 癌胚抗原(carcinoembryonicantigen,CEA) CEA为相对分子质量为180 000 ~200 000 的糖蛋白,属于免疫球蛋白超家族,其表达量与肿瘤发生率呈正相关[10]。在正常组织中,CEA 及其家族成员可表达于胃肠道上皮,健康人血清中CEA 水平极低;但在多种上皮细胞癌中过量表达,通过增强肿瘤细胞的侵袭及增殖能力,使其易于扩散转移,血清检测CEA水平也明显升高,一般超过20 μg / L,提示有消化道肿瘤,结直肠腺癌患者尤其显著,术前血清CEA 阳性率为44.07%[11]。约30% ~60%胰腺癌患者CEA表达阳性,其血清检测水平也明显升高[12]。因此,CEA 可作为治疗消化道肿瘤的理想靶点。
2.3 人类表皮生长因子受体2(human epidermal growth factor receptor 2,HER2) HER2 又称 ErbB-2 /neu,位于17 号染色体长臂(17q12-21.32),可编码相对分子质量为185 000 的具有酪氨酸激酶活性的跨膜受体蛋白。生理情况下,通过受体与配体相结合,促使HER2 二聚化,激活下游MAPK、PI3K 等信号通路,促进细胞增殖、防止细胞凋亡[13]。当细胞表面HER2过表达,细胞会发生过度增殖及恶性转移。因此,通过阻断活化的HER2 信号通路,抑制肿瘤细胞增殖及启动凋亡途径,促进肿瘤细胞程序性死亡。约20%的原发性乳腺浸润性导管癌有HER2 过表达[14];雄激素非依赖性前列腺癌(androgen independent prostate cancer,AIPC)患者HER2 表达阳性率高于良性前列腺增生和雄激素依赖性前列腺癌患者[15];20%~60%的胰腺导管腺癌(pancreatic ductal adenocarcinoma,PDAC)患者中也有阳性表达[16]。因此,HER2 可作为治疗表达阳性率较高肿瘤的理想靶点。
2.4 前列腺干细胞抗原(prostate stem cell antigen,PSCA) PSCA 是一种前列腺特异性基因,可编码123个氨基酸,通过GPI 锚定在细胞表面,与Thy-1 / Ly-6家族中干细胞抗原-2 具有30%同源性[17]。生理条件下,PSCA 在前列腺中表达;病理情况下,PSCA 可过表达于多种癌症组织,包括前列腺癌、胃癌、胰腺癌、膀胱癌等。其可激活PI3K / AKT 信号通路,促进癌细胞增殖分化;而沉默其表达可抑制细胞增殖[18]。有研究显示,超过80%的前列腺癌中PSCA 呈过表达;胰腺癌中PSCA 阳性率约77.1%;慢性胰腺炎组织中PSCA 阳性表达率为31.3%[19-20]。因此,PSCA的高表达可作为一个新的治疗靶点和评价肿瘤发生发展预后的指标。
2.5 黏蛋白(mucin-1,MUC-1) MUC-1 是一种相对分子质量大于200 000 的Ⅰ型跨膜糖蛋白。生理条件下,MUC-1 极性分布于呼吸道、胃肠道、泌尿生殖道、乳腺等组织的上皮细胞近管或腺腔面,有促进细胞生长、分化、信号传导及保护和润滑上皮细胞等功能。但在多种上皮细胞瘤中,MUC-1 高表达且非极性分布于整个胞膜甚至胞质中。其中胰腺癌阳性率约 90%[21],胃癌阳性表达率约 65%[22]。其致病机制主要包括:MUC-1 与细胞间黏附分子-1(intercellular cell adhesion molecule-1,ICAM-1)或 E-选择素(e-selectin)受体结合,导致肿瘤细胞与血管内皮细胞黏附,癌细胞可穿过血管内皮发生远处转移;过表达MUC-1 可抑制E-钙黏蛋白介导的细胞间相互作用,降低循环肿瘤细胞对血管内皮的黏附,因此易发生脱落并随血液循环向远处侵袭转移;MUC-1细胞外结构域可维持肿瘤失巢凋亡细胞的表面完整性、阻止失巢起始分子活化及防止肿瘤细胞凋亡等[23-24]。因此,MUC-1 是一种高表达且在癌症的发生、发展、扩散及抗凋亡中发挥重要作用的蛋白,可作为癌症新型免疫疗法的靶点。
2.6 CD24 CD24 是通过GPI 锚定的膜蛋白,编码27 ~30 个氨基酸,人的CD24 基因位于染色体6q21上[25]。生理条件下,CD24 多分布于祖细胞和代谢活跃的细胞上,在终末分化细胞中表达较低。通过传导细胞间信号来调节细胞的增殖分化、机体自身免疫及诱导成熟细胞凋亡等。病理情况下,其在多种肿瘤组织中广泛表达,可加速肿瘤生长、侵袭和转移。研究发现,174 例卵巢癌中,浆液性腺癌(87.5%)和浆液性乳头状癌(84.6%)患者的阳性表达率显著高于其他组织学亚型,87.9%的淋巴结转移患者中发现有CD24 的表达,无淋巴结转移的患者中有60.7%呈CD24 的阳性表达[26]。因此,作为阳性表达率较高的肿瘤抗原,CD24 可作为表达阳性肿瘤的理想靶点。
2.7 Claudin18.2(CLDN18.2) CLDN18.2 是一种跨膜蛋白,其正常表达对维持细胞间紧密连接起重要作用[27]。CLDNs 蛋白的生理功能有:维持细胞极性和上皮细胞渗透压、参与细胞间信号转导及形成天然屏障防御病原体入侵等[28]。CLDN18.2 在正常组织中表达受限,仅表达于分化的胃黏膜膜上皮细胞。但在多种恶性肿瘤中,CLDN18.2 呈高表达,尤其是消化道肿瘤:77%原发性胃腺癌表达阳性;78%原发性食管腺癌表达阳性;80%胰腺癌表达阳性[29]。根据其相对较高的特异性表达,CLDN18.2 可作为治疗消化道肿瘤的理想靶点。
3 实体瘤CAR-T 治疗方案的设计
由于实体瘤细胞具有高异质性且缺乏TSAs,导致患有同种肿瘤的不同患者体内抗原表达有差异,且肿瘤细胞表面高表达的抗原在正常组织也有少量或微量表达,造成CAR-T 细胞疗法用于实体瘤时产生严重的抗原逃逸现象和脱靶效应。如MORGAN等[30]研究将HER2 作为靶点治疗患有结肠癌合并肝肺部转移的患者时,在输注1 × 1010个CAR-T 细胞后15 min 内,患者出现呼吸窘迫,胸部X 线显示肺部浸润明显,于治疗后第5 天死亡,其血清样本显示,IFNγ、TNF-α、GM-CSF、IL-6 和 IL-10 等细胞因子水平显著增加,证明患者在接受治疗后,由于CAR-T细胞识别正常肺上皮细胞上低水平表达的HER2 抗原,诱发了强烈的细胞因子风暴。因此,针对CAR-T用于实体瘤时产生的严重不良反应,应针对不同的患者设计不同的方案,以降低副反应的发生。
3.1 通过串联结构减轻抗原逃逸现象 HEGDE等[31]将胶质母细胞瘤作为研究对象,设计出串联HER2-scFv 和IL13Rα2-IL13 突变蛋白的二代CART(TanCAR-T)细胞,结果表明,当TanCAR-T 细胞同时靶向HER2 和IL13Rα2 这两种抗原时,T 细胞会发生超强活化,与对照组相比,T 细胞的活性更持久且不易耗竭,显示出了更有优势的抗肿瘤效果。动物实验也发现,TanCAR-T 细胞治疗后小鼠寿命明显延长。同时还发现,复发的肿瘤细胞表面这两种靶抗原缺失,证明串联的CAR-T 结构可减轻肿瘤抗原逃逸现象,防止肿瘤细胞逃避免疫监视。有研究针对卵巢癌细胞表面FOLR1 和MSLN 的两种抗原,成功构建了四代CAR-T 细胞,同时与单靶点CAR-T 细胞相比,发现串联CAR-T 的体内外杀伤效能均强于单靶点组,能分泌更高水平的细胞因子,进一步为串联CAR-T减轻抗原逃逸等现象提供了实验依据[32]。
3.2 通过并联结构减轻脱靶效应 ZHANG 等[33]选择胰腺癌作为研究对象,将T 细胞表面同时表达CEA-scFv 和MSLN-scFv 双嵌合抗原受体,即并联CAR-T(dCAR-T)。T 细胞仅在同时靶向CEA 和MSLN 两种肿瘤抗原情况下被完全激活。通过体内外实验结果均表明,dCAR-T 细胞可精确定位于肿瘤部位,并对肿瘤细胞产生高细胞毒性,同时不损伤相邻组织。dCAR-T 细胞提高了其精准杀伤癌细胞的作用和治疗安全性,在较大程度避免了CAR-T 治疗中“脱靶效应”的发生。KLOSS 等[34]选用前列腺癌细胞表面特异性较高的靶点,前列腺特异性膜抗原(prostrate specific membrane antigen,PSMA)和PSCA,设计了同样的并联结构。通过前列腺癌小鼠模型中证明,dCAR-T 细胞能根除PSCA+PSMA+肿瘤细胞,发挥强大且有效的杀伤作用,但对任一抗原阴性的肿瘤细胞则无治疗效果。因此,并联设计不仅提高了靶向肿瘤细胞的精准性,减轻脱靶效应的发生,还提高了抗肿瘤活性。上述研究表明,基于肿瘤免疫疗法,用两种不同的TAA 来调节dCAR-T 细胞活性的组合疗法具有较强的应用前景。
3.3 通过逃避免疫抑制检查点增强体内抗肿瘤功效RAFIQ 等[35]设计的 CAR-T 细胞靶向递送 PD-1 阻断性scFv,并验证体内抗肿瘤功效。通过自分泌或旁分泌释放的PD-1 scFv 与肿瘤细胞表面PD-L1 结合,保护T 细胞表面PD-1 信号分子免受肿瘤细胞的抑制,提高CAR-T 细胞抗肿瘤活性。动物实验结果证明,分泌PD-1scFv 的CAR-T 细胞可使荷瘤小鼠长期存活,并发现小鼠骨髓中有长期存活的CAR-T 细胞,当肿瘤复发时可再次启动抗肿瘤免疫应答;用PD-1scFv 和PD-1mAb 分别处理腹膜癌小鼠模型发现,PD-1scFv 仅在腹膜腔分布,证明其可准确定位于TME 来增强CAR-T 细胞的抗肿瘤活性,在外周血清中未检测到其表达,在小鼠全身均可检测到PD-1mAb。因此,阻断PD-1 或其受体可防止T 细胞的活性被抑制,增强其杀伤能力,串联PD-1scFv 的CAR-T治疗可能是代替PD-1 抗体药物更安全有效的治疗方案。CHEN 等[36]设计了CAR-T 细胞表面过表达PD-1 DNR(PD-1 dominant negative receptor),可充当“诱饵受体”以结合和阻断PD-L1/2 的抑制信号,与抗PD-1 抗体比较发现,通过基因改造的CAR-T 细胞提供的PD-1 阻断信号是一种可持续抑制效果,而抗PD-1 抗体的功效则受肿瘤渗透率低,半衰期短以及非特异性毒性限制。RABELLO 等[37]针对转移性肾透明细胞癌,设计了以人抗碳酸酐酶Ⅸ(carbo-nic anhydrase Ⅸ,CAⅨ)为靶点的CAR-T 细胞,并将其设计为分泌人抗PD-L1 的抗体。结果发现与单独的抗CAIX CAR-T 细胞相比,肿瘤的生长减少了5 倍,肿瘤重量减少了50% ~80%。上述研究结果表明,这些创新点具有较大潜力。
3.4 改变肿瘤微环境增强T 细胞的抗肿瘤活性ADACHI 等[38]通过两个 2A 肽将 IL-7 和 CCL19 串联,并与CAR-T 细胞连接,形成可分泌IL-7 和CCL19的CAR-T 细胞,用以模拟T 细胞区网状细胞的功能。T 细胞区的成纤维网状细胞所产生的IL-7 和CCL 19 对淋巴器官中T 细胞区的形成和维持至关重要。通过两者共同作用,将T 细胞和DC 细胞募集至肿瘤区域。实验结果显示,1 × 106个串联CAR-T 细胞的疗效相当于4 × 106个传统的CAR-T 细胞,抗肿瘤能力有明显提高,证明IL-7 和CCL19 具有可增强 CAR-T 细胞抗肿瘤的能力。HOMBACH 等[39]将CD30-scFv 与抗CEA CAR-T 细胞相串联,证明C15A3癌细胞与抗CD30/CEA CAR-T 细胞和抗CEA CAR-T细胞共培养时,前者的抗肿瘤作用更显著,进一步证明串联CD30-scFv 的CAR-T 细胞在靶向其他TAAs 时,均可提高CAR-T 细胞消除癌细胞的效率。KONERU 等[40]设计了针对卵巢癌细胞表面MUC-16ecto 抗原为靶点且同时分泌IL-12 的CAR-T 细胞,通过体内外实验均证明IL-12 的分泌可增强IFNγ 分泌能力和抗肿瘤功效。
4 临床研究进展
由于实体肿瘤靶点多样且具有高度异质性,因此,在治疗实体瘤时需要选取阳性表达率相对较高的抗原作为靶点。有临床试验数据显示,经临床Ⅰ期CAR-T 治疗,10 例CEA 阳性难治复发性结直肠癌患者,治疗后有7 例疗效评价为病情稳定,其中2 例病情稳定保持时间超过30 周,另有2 例影像学报告显示,肿瘤病灶及代谢均减小,大部分患者血清CEA含量明显降低[41]。BEATTY 等[42]对 6 名难治性转移性PDAC 患者进行靶向间皮素的CAR-T 治疗,结果显示,2 例患者病情稳定,无进展生存时间分别为3.8 和5.4 个月;3 例患者肿瘤病变的代谢活性体积保持稳定;1 例患者活检证实MESO 表达率下降69.2%;接受治疗的患者血液中均检测到瞬时CAR表达,产生了新的免疫球蛋白IgG。
通过国际临床试验注册网站(ClinicalTrials.gov)检索发现,有413 项已注册的CAR-T 相关临床试验。其中实体瘤共29 项,主要靶点包括EGFR、HER2、Claudin18.2、Mesothelin、PSCA、MUC-1 等。正在开展的实体瘤CAR-T 疗法临床试验情况见表1。
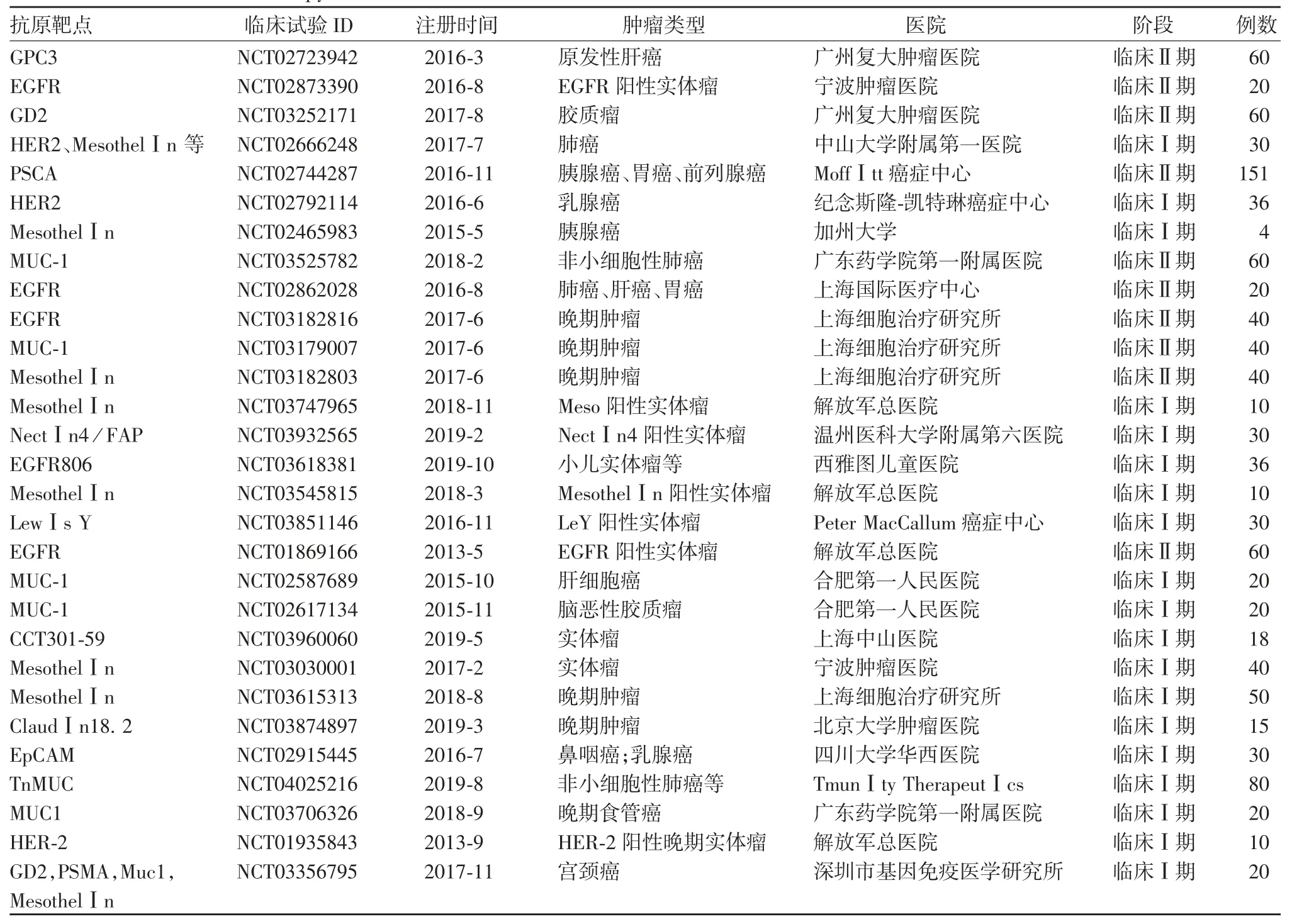
表1 CAR-T 靶向实体瘤抗原治疗在国际临床试验注册网站的注册情况Tab.1 License of CAR-T therapy in international websites of clinical trials
5 小结与展望
尽管CAR-T 细胞治疗血液瘤效果显著,由于实体瘤缺乏特异性靶点、肿瘤抗原逃逸及TME 抑制等复杂因素,使其疗效受限。因此,通过寻找特异性较高的抗原作为治疗靶点、优化CAR-T 结构及改善TME 抑制等方式来提高T 细胞靶向肿瘤的精准性、增强抗肿瘤疗效。另外,CAR-T 细胞联合溶瘤病毒的组合治疗策略、联合PD-1 / PD-L1 单抗用药及联合放化疗等治疗手段临床试验也在相继开展。随着对肿瘤免疫逃逸机制的深入研究及CAR-T 治疗策略不断完善,有望进一步提高CAR-T 细胞迁移浸润并杀伤肿瘤组织的能力,进而研发出更加安全有效及高效的实体瘤临床治疗策略。
猜你喜欢
保健医苑(2022年5期)2022-06-10 07:46:38
中老年保健(2021年3期)2021-12-03 02:32:25
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
中国生殖健康(2020年7期)2020-12-10 07:48:51
西南医科大学学报(2015年1期)2015-08-22 13:01:46
医学研究杂志(2015年6期)2015-07-01 17:41:11
医学研究杂志(2015年7期)2015-06-22 11:01:36
医学研究杂志(2015年7期)2015-06-22 11:01:01
癌变·畸变·突变(2015年3期)2015-02-27 06:15:07
