
牛病毒性腹泻病毒非结构蛋白p7 的克隆和原核表达纯化
2021-06-17 02:59付强郭妍婷杨莉史慧君
中国畜禽种业 2021年5期
付强 郭妍婷 杨莉 史慧君
(新疆农业大学动物医学学院 830052)
牛病毒性腹泻病毒(Bovine Viral Diarrhea Virus,BVDV)属于黄病毒科(Flaviviridae)、瘟病毒属(Pestivirus),主要感染牛、羊、骆驼等家畜和野生动物[1],是牛病毒性腹泻-黏膜病(Bovine Viral Diarrhea-Mucosal Disease,BVD-MD)的主要病原之一[2]。该病毒感染成年动物后常呈现隐性感染,一般不表现明显的临床症状,而幼畜感染后常造成腹泻、高烧、白细胞减少、黏膜糜烂甚至脱落等症状,感染孕畜后造成流产、产死胎和畸形胎、繁殖障碍等[3,4]。尤其是非致细胞病变型(Non-cytopathogenic Effect,NCP)BVDV 感染妊娠早期母畜,造成持续性感染(Persistent Infection,PI)犊牛的出现,PI 犊牛一旦再次感染CP 型BVDV 后表现出严重的腹泻黏膜病,最终因脱水和代谢性酸中毒等致死率高达90%,而且PI 犊牛始终向外排毒,成为污染整个牛群的重要传染源[5,6]。
BVDV 非结构p7 蛋白在病毒复制过程中起到重要的作用。有研究报道,非结构蛋白p7 翻译后折叠并加工成六聚体形式,转运至细胞膜上,组成离子孔道,所以p7 蛋白属于离子孔道蛋白家族,主要参与介导一些离子(Na+、Ca2+、K+、H+等)运输,而且对病毒进入细胞及子代病毒在细胞中成熟产生影响[7-9]。本研究开展BVDV 非结构蛋白p7 的基因克隆及原核表达纯化,以期获得p7 蛋白,为后续构建p7 的多克隆和单克隆抗体提供重要的材料和平台。
1 材料和方法
1.1 细菌、载体和cDNA
BL21(DE3)PLySs 大肠杆菌感受态细胞购自北京索莱宝科技有限公司;pCold-MBP 载体购自武汉淼灵生物科技有限公司;BVDV 毒株NADL 的cDNA 保存于新疆农业大学动物医学学院动物病毒实验室。
1.2 主要试剂和仪器
考马斯亮蓝快速染色液、透析袋和5×蛋白上样缓冲液购自北京索莱宝科技有限公司;Kpn I 和BamH I 购自NEB 公司;Taq PCR Mastermix、T4 Ligase、琼脂糖凝胶DNA 回收试剂盒和普通质粒提取试剂盒购自天根生化科技(北京)有限公司;Ni-TED 琼脂糖树脂、NaH2PO4、NaCl、咪唑imidazole、琼脂糖和异丙基-b-D-硫代半乳糖苷IPTG 购自生工生物工程(上海)股份有限公司;低温高速台式离心机Micro 21R 购自美国Thermo 公司;C1000 PCR 仪购自美国BioRad 公司;DYCP-31DN型核酸琼脂糖水平电泳仪购自北京六一生物科技有限公司。
1.3 p7 基因克隆及测序分析
根据GenBank 数据库中BVDV NADL(NC_001461.1)基因组序列中p7 基因,设计扩增p7 基因引物为p7-F:5’CGGggtaccATGTCCCAGTATGGGGCAGGTG 3’;p7-R:5’ CG CggatccAGCCT TTGCCATCCCTCCAAC 3’,其中ggtacc 和ggatcc分别为Kpn I 和BamH I 酶切位点。以BVDV NADL cDNA 为模版,以p7-F 和p7-R 为引物,PCR 扩增p7 基因,PCR 反应体系见表1,PCR 反应条件为95℃5min;95℃30s、60℃30s、72℃30s,35 个循环;72℃10min;4℃∞。使用琼脂糖凝胶电泳检测PCR 结果,并使用琼脂糖凝胶DNA 回收试剂盒回收p7 基因,将p7 基因送至苏州金唯智生物科技有限公司进行测序分析。

表1 PCR 反应体系
1.4 pCold-MBP-p7 原核表达载体构建
使用Kpn I 和BamH I 对p7 基因和pCold-MBP 质粒进行酶切,酶切体系见表2,酶切反应条件为37℃4 h。酶切反应完成后使用琼脂糖凝胶电泳进行检测,并使用琼脂糖凝胶DNA回收试剂盒将酶切后的p7 基因和pCold-MBP 载体回收,使用T4 DNA Ligase 将p7 基因和pCold-MBP 载体连接,连接反应条件见表3,连接反应条件为16℃过夜;将连接产物热激转化至BL21(DE3)PLySs 大肠杆菌感受态细胞,涂布到含有氨苄西林的琼脂平板中,37℃培养过夜,次日使用PCR 鉴定单克隆菌落,并将阳性克隆送至苏州金唯智生物科技有限公司进行测序鉴定。

表2 酶切反应体系
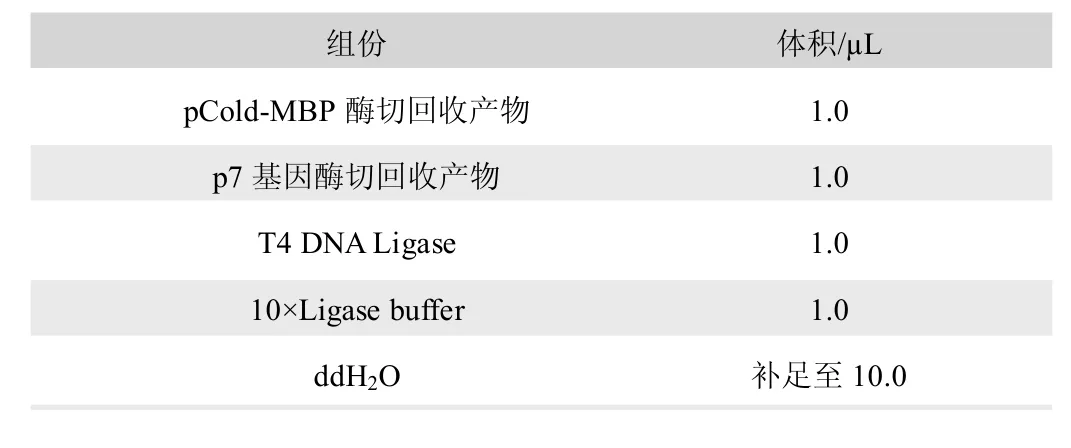
表3 连接反应体系
1.5 IPTG 诱导条件优化
将pCold-MBP-p7 单克隆菌落接种至LB 肉汤培养基中扩繁,扩繁完成后以体积比1:100 比例接种至新的LB 肉汤培养基中,置于37℃200rpm 振荡培养,待OD600 nm 为0.4~0.6时,分别加入终浓度0、0.6、0.8、1.0、1.2 mM IPTG,16℃条件下200rpm 振荡过夜诱导表达;次日分别取1ml 培养物,8000rpm 离心5min 后弃掉上清,加入100μL 1×蛋白上样缓冲液,使用SDS-PAGE 电泳分离蛋白质,并使用考马斯亮蓝染色套装(染色液+脱色液)进行染色和脱色,具体染色和脱色步骤见试剂盒说明书。
1.6 pCold-MBP-p7 原核表达载体诱导表达和纯化
pCold-MBP-p7 单克隆菌落扩繁步骤同上,扩繁后以体积比1:100 比例接种菌液至新的LB 肉汤培养基中,置于37℃200rpm 振荡培养,待OD600nm为0.4~0.6 时,加入终浓度为1.2 mM IPTG,置于16℃低温过夜诱导表达,次日离心收集菌体沉淀,加入10ml 裂解液(50 mM NaH2PO4,500mM NaCl,20mM imidazole,调节pH 至8.0)重悬沉淀,使用细胞超声破碎仪(15s ON,15s OFF)进行超声破碎至液体清亮。8,000rpm 离心10min 去除细胞碎片,填加到Ni-TED 琼脂糖树脂中,4℃孵育4h 后使用3 倍柱体积的洗涤缓冲液(50mM NaH2PO4,500mM NaCl,40mM imidazole,调节pH 至8.0)进行洗涤,加入洗脱缓冲液(50mM NaH2PO4,300mM NaCl,250mM imidazole,调节pH 至8.0)进行4 次洗脱,收集洗脱溶液,进行SDS-PAGE电泳并使用考马斯亮蓝溶液染色进行观察。
1.7 蛋白透析及检测
将透析袋裁剪成适当长度,置于2% NaHCO3和1mM EDTA 溶液中煮沸10min;使用蒸馏水清晰透析袋后,使用1mM EDTA 溶液煮沸透析袋10min,待冷却后灌装洗脱蛋白溶液,置于100 倍体积的磷酸盐缓冲液(20mM,pH 7.0)放置于4℃下渗透过夜,次日转移剩余蛋白溶液至15ml 离心管,取少量蛋白溶液进行SDS-PAGE 电泳,使用考马斯亮蓝溶液染色进行观察,其余置于-80℃为后续使用。
2 结果
2.1 p7 基因扩增及pCold-MBP-p7 原核表达载体构建
以BVDV NADL cDNA 为模版,使用PCR 扩增p7 基因。如图1 所示,PCR 产物大小约为225bp,与p7 基因大小一致;将p7 基因克隆至pCold-MBP 载体后,获得pCold-MBP-p7 原核表达载体,质粒图谱见图2。
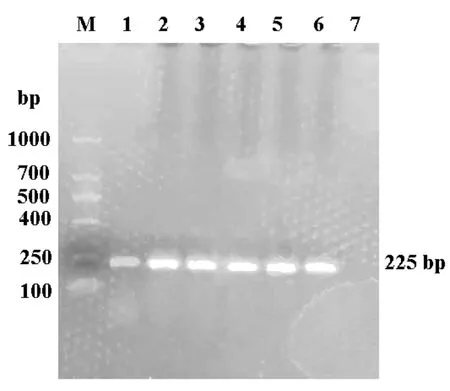
图1 PCR 扩增p7 基因结果

图2 pCold-MBP-p7 原核表达载体图谱
2.2 IPTG 诱导条件优化
加入不同浓度的IPTG 后低温过夜诱导,次日收集菌体并使用SDS-PAGE 进行检测。结果如图3 所示,使用1.2mM IPTG 诱导后能蛋白表达量最高,所以后续试验使用1.2mM IPTG 作为诱导条件。

图3 IPTG 诱导浓度优化
2.3 pCold-MBP-p7 原核表达和蛋白纯化
使用IPTG 诱导pCold-MBP-p7 表达过夜,收集菌体后破碎,使用Ni-TED 琼脂糖树脂进行纯化。结果如图4 所示,MBP-p7 融合蛋白成功表达,约49kDa;经过4 次洗脱液洗脱后大部蛋白已从琼脂糖树脂洗脱下来,得到杂带较少的MBPp7 融合蛋白。

图4 MBP-p7 蛋白纯化
2.4 MBP-p7 蛋白透析
使用透析袋对MBP-p7 蛋白进行透析,完成后使用SDS-PAGE进行电泳,使用考马斯亮蓝染色进行鉴定。结果如图5 所示,透析后MBP-p7 蛋白条带较为单一,表明MBP-p7 蛋白纯度高。

图5 MBP-p7 蛋白透析后鉴定
3 讨论
黄病毒科非结构蛋白p7 经翻译后加工聚集并转运至细胞膜上形成六聚体离子孔道形式,参与到离子跨膜运输、病毒侵入和释放等过程,对病毒复制起到重要作用[10]。而目前关于p7 蛋白如何进行聚集,在什么时间p7 蛋白开始聚集尚不清楚,本课题组正在开展BVDV p7 形成离子孔道的机制研究,而目前尚未见BVDV p7 蛋白相关的抗体销售,开展本试验表达纯化p7 蛋白,拟为后续p7 蛋白的多克隆抗体及单克隆抗体研发提供重要的材料。
本试验使用pCold-MBP 原核表达载体进行诱导表达纯化,其中pCold 属于冷休克基因CSPA 启动子,在较低温度下即可诱导蛋白表达,进而降低蛋白诱导过程中因温度过高造成的蛋白聚集、变性、形成难溶物等情况发生[11];MBP 蛋白标签是一种麦芽糖结合蛋白标签,分子量约42.5 kDa,一般情况下将MBP 蛋白融合在目的蛋白的N 端,有利于减少目的蛋白的降解,提高蛋白的可溶性,维持蛋白结构稳定性,减少纯化难度[12]。本试验使用pCold 实现低温高效诱导表达,使用MBP 蛋白标签实现维持蛋白稳定性的作用。
猜你喜欢
环球时报(2022-09-20)2022-09-20
湖南畜牧兽医(2021年6期)2022-01-24
临床与实验病理学杂志(2021年10期)2021-12-13
食品安全导刊(2021年21期)2021-08-30
商品与质量(2021年31期)2021-08-20
猪业科学(2021年5期)2021-06-02
检验医学(2021年4期)2021-04-28
中国科技纵横(2021年24期)2021-03-02
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
