
从乳酸铵发酵液提取乳酸的技术研究
2021-06-11 04:07张国宣侯玉梅
河南化工 2021年5期
王 浩 , 郭 廷 , 张国宣 , 侯玉梅
(河南金丹乳酸科技股份有限公司,河南 郸城 477150)
乳酸(HLAC)是三大有机酸之一,未来的发酵有机酸市场中乳酸的需求量将超过现有的柠檬酸,而跃居第一位[1]。乳酸发酵液的成分复杂,并且因原料和发酵工艺的不同而各不相同。除乳酸外,发酵液中还包括菌体、残糖、蛋白质、色素、胶体、有机杂酸、无机盐等多种杂质[2]。总的来说,它们来源于原材料、未消耗的营养盐或发酵的中间副产物,难度比较大,从乳酸铵发酵液中提取乳酸更是比较困难。从发酵液中分离和纯化产物的“下游工程”,即乳酸提取技术的现状来看,难以跟上当今乳酸生产技术的发展步伐[3]。因此,如何完善和改进现有的乳酸发酵以及发酵后提取和精制工艺,寻求更为经济、有效的分离提取方法,以提高产品的质量和产品的收率,降低生产成本,适应大规模生产的需要,已成为一个重要的课题。国内外萃取技术研究成效显著,通过萃取分离技术应用可以提高产品纯度,降低产品色度、还原糖、醚不溶物[4]。国内报道乳酸铵发酵液提纯采取脱色、超滤除蛋白、强酸性阳树脂和强碱性阴树脂进行脱盐转酸,收率偏低,仅65.8%[5]。本文就乳酸铵发酵液提取乳酸的方法作了深入探讨,开拓了非钙盐法发酵生产乳酸的新思路,具有高效、处理量大、操作简单、易于放大等优点,为铵盐法发酵生产乳酸产业化提供技术支撑[6]。
1 实验材料与仪器
发酵液(乳酸铵溶液),由发酵实验室提供,乳酸浓度22%,残还原糖0.4%,过50 nm陶瓷膜除去菌体及固形物;萃取剂;生物传感分析仪;紫外分光光度仪 TU-1810;ICP-AES(电感耦合等离子体原子发射光谱);125 mL分液漏斗,500 mL分液漏斗、分液漏斗架;数显恒温水浴锅(带磁力搅拌器);旋转蒸发仪;循环水真空泵;层析柱。
2 萃取工艺基本流程
本实验萃取工艺基本流程:①通过逆流萃取将乳酸从酸化料中萃取进入有机相,用纯水在常温下进行小相比逆流水洗;②用纯水逆流反萃,使有机相中的乳酸再次进入到水相中,得到乳酸溶液;③进行脱脂棉脱油,活性炭脱色,浓缩得到产品。将得到的产品乳酸按GB 1886.173—2016食品安全国家标准食品添加剂乳酸国家标准检测。
实验由以下5部分组成:①萃取剂选择,萃取工艺条件的确定;②乳酸的萃取提取,形成含有乳酸的负载有机相和带有很多杂质的萃余液;③水洗,用去离子水洗涤负载有机相,去除色素和其他离子;④反萃,将乳酸从负载有机相中分离出来形成乳酸溶液和空白有机相;⑤将反萃出来的乳酸溶液通过脱脂棉脱除乳酸稀溶液中残余的油脂。流程图见图1。
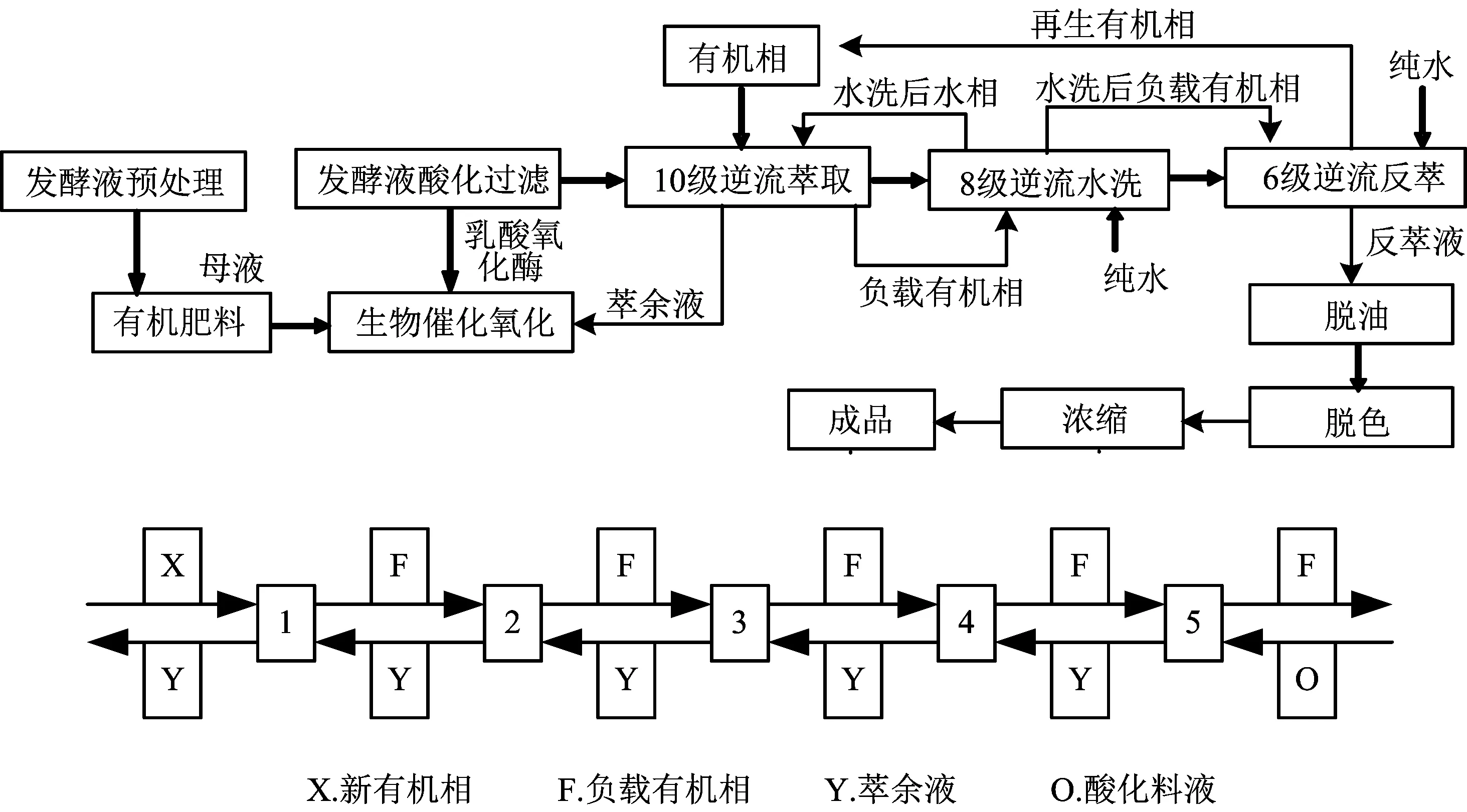
图1 多级逆流萃取示意图
2.1 基本原理
在溶剂萃取中,被萃取物质乳酸在水相和有机相中进行分配,其行为与有机相的性质相关。本实验用到的萃取剂为物理性萃取剂,是相似相溶的原理,有机相物质对有机物分子态乳酸有一定的溶解度,对盐类这些极性物质溶解性较小,因此在萃取过程中水相中的分子态乳酸会进入到有机相中,而水相中的硫酸铵、硫酸氢铵、硫酸钙等极性物质则保留在水相中。同理,可用纯水再次将有机相中的乳酸反萃到水相中,然后将得到的反萃液进行后续处理得到乳酸成品。
2.2 发酵液酸化终点确定
由于乳酸发酵后以乳酸铵盐类的形式存在于发酵液中,而萃取剂对乳酸的萃取是以分子态的方式进行,因此必须将发酵液中的乳酸根离子转化为乳酸分子,才能进行下一步的反应,所以萃取前必须对发酵液进行酸化处理。
酸化的终点控制:根据乳酸的电离常数为1.4×10-4,根据以下公式:
[H+]×[LAC-]÷[HLAC]=1.4×10-4
[LAC-]÷[HLAC]≈1∶100 (使分子态乳酸比例为99%)
则
[H+]=1.4×10-2
则酸解终点pH值可计算为:
pH值=-lg[H+]=-lg[1.4×10-2]=1.85
酸解料的pH值应控制在1.85以下,酸化终点可控制在1.5~1.8。
2.3 萃取、反萃取温度
根据乳酸的特点和萃取剂的性质选择了萃取和反萃取的温度。高的萃取温度会带来更快的传质速度,可以更快地达到萃取平衡,也可以带来更高的分配比;但高的温度会有更多的能源消耗,更多的萃取剂挥发性损失,因此需要选择一个折中的温度,实验中选择了60 ℃。
乳酸随温度的降低在有机相中的溶解度减小,因此选择低温反萃,温度越低则一次反萃率越高,但是低温降低了传质速度,在冷却过程中也会带来大量能耗,因此没有必要将温度设定在常温以下,实验选择常温25 ℃进行反萃。
2.4 萃取剂饱和容量与分配比
根据萃取剂的性质,萃取剂的饱和容量与乳酸在萃取剂中溶解度密切相关,萃取剂的分配比与相比关系不大。在饱和容量与分配比的测试中采取的有机相与水相的比例为3∶1,在60 ℃下,采用单级错流萃取的方式,每次将水相移除,换上新的水相,直至从分液漏斗中流出的水相浓度与酸化料液浓度相同或接近为止。实验结果如表1所示。

表1 萃取剂饱和容量与分配比测试结果 %
饱和容量用X(g/L)表示:
分配比用D表示:
式中:n为萃取次数;c为酸化料浓度;cn为n次萃余液浓度;经过计算此萃取剂的饱和容量X=9.37 g/L,分配比D=0.45。
2.5 酸化料的浓度
酸化料的浓度可根据萃取剂的分配比、有机相饱和容量、有机相饱和时水相乳酸的浓度确定。
在60 ℃下萃取剂的分配比为0.45,饱和容量为9.37 g/L,萃取过程中在有机相饱和时,水相质量百分比为9.37/0.45=20.8,因此酸化过滤液中乳酸质量百分比不应低于20.8%,酸化料的乳酸质量百分比控制在25%左右,以避免由酸化料中乳酸质量百分比不足造成的萃取剂利用率低的问题,质量百分比过高会使溶液黏度过大不利于萃取。
2.6 萃取剂相比确定
萃取剂相比可根据酸化料的乳酸质量百分比和有机相的饱和溶液浓度来确定。
实验酸化料的乳酸质量百分比为22%,则萃取的相比应大于22/9.37=2.34,以避免由于相比过小造成有机相饱和,水相中乳酸萃取不完全造成萃余液残余乳酸浓度过高的问题,试验采用2.5的相比是合适的。
2.7 萃取级数确定
萃取级数按以下公式计算:
n+1=[lg(EA+qA-1)-lgqA]÷lgEA
式中:n,萃取级数;EA,相比×分配比;qA,逆流萃取后乳酸残留率。
实验中相比为2.5,分配比为0.45,要求乳酸残留率在1%以下,qA=0.01,经计算取n=10,即采取10级逆流萃取。
反萃采用负载有机相与水相比例为2.5∶1,由于反萃在常温下进行,其在水中与有机相中分配比高于1/0.45,其分配比在2.5左右,因此相比于逆流萃取可适当减少级数,实验采取的是6级常温逆流反萃。
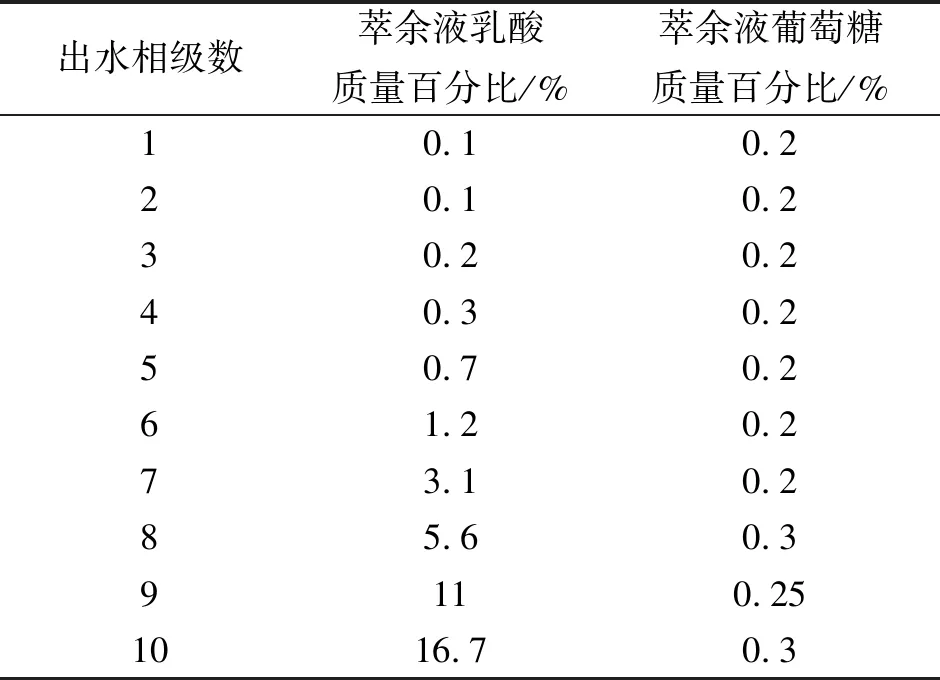
表2 10级逆流萃取平衡后各级参数分析

表3 6级逆流反萃平衡后各级参数分析 %
2.8 脱油
反萃得到的稀乳酸中存在少量的萃取剂,既影响后续处理工艺,也影响最终产品的质量,因此必须对反萃得到的稀乳酸溶液进行脱油处理。方法如下:将剪碎的脱脂棉装一个6 cm×100 cm的层析柱,径高比为10,然后将反萃液加入层析柱中以一定的线速度(1.67 m/s)流出。结果表明经过脱油处理,残余在反萃得到的稀乳酸中的油脂可基本被完全脱除。
2.9 脱色
反萃得到的稀乳酸经过脱油后含有大量酸性色素,呈亮黄色,因此需要活性炭脱色。本实验采用颗粒活性炭固定床来进行,试验过程如下:
2.9.1活性炭的预处理
颗粒活性炭→纯水浸泡4 h→5%烧碱溶液浸泡12 h→纯水洗pH值接近中性→5%盐酸溶液浸泡12 h→用水洗至甲基橙试剂变黄。
2.9.2脱色
将处理好的颗粒活性炭装一个6 cm×100 cm的层析柱,径高比为10,然后将反萃得到的脱油后的稀乳酸加入层析柱中以一定的线速度(1.67m/s)流出。
2.9.3脱色结果
反萃液在脱色后溶液透亮,经紫外分光光度计测其透光率显示其在可见光区全波段透光率在99%以上。如图2所示。

图2 反萃液脱色前后透光率全谱扫描图
2.10 浓缩
浓缩采用减压蒸馏法,控制温度在70 ℃,真空度-0.99 MPa,时间1 h,得到样品乳酸浓度约90%。
2.11 水洗
由萃取法从乳酸铵经过萃取反萃得到的成品中,还有一定的铵离子及其它非乳酸离子存在,影响了所制得的乳酸成品的质量。通过离子交换树脂可以去除离子,但树脂对铵离子的脱除效率较低,增加了树脂的负荷,不经济。因此,经过反复试验,利用物质的极性不同在萃取和反萃之间多级水洗,将铵及其它离子洗脱,以达到去除杂质离子的目的。
水洗相的加入也是存在弊端的,它降低了负载有机相的质量百分比(在负载有机相与水相比为9∶1时,降低2个浓度左右),从而降低了反萃液的质量百分比,使有机相的运行效率下降约22%。
水洗共做了2组,一组是5级的水洗,一组是8级的水洗。其中5级水洗最后得到成品分析不合格,8级水洗得到的成品分析合格,并且反萃液加碱调pH值=12,已经闻不到氨味。
2.12 指标检测结果
样品按GB 1886.173—2016食品安全国家标准食品添加剂乳酸标准检测,通过以上工艺条件10批次实现,所得乳酸产品主要指标检测结果如表4所示。

表4 乳酸产品指标
样品经ICP-AES检测其金属离子含量如表5所示。
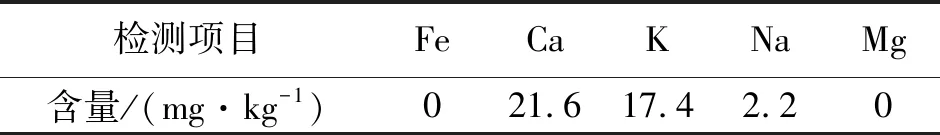
表5 产品金属离子含量
从表5可以看出,利用萃取与反萃技术从复杂体系中提取乳酸是可行的,是一种提取乳酸的新方法。
3 试验结果
通过试验的工艺参数得到优化确定:酸化终点可控制在pH值1.5~1.8,萃取温度60 ℃,反萃取温度25 ℃;有机相与水相的比例为3∶1,萃取剂的饱和容量X=9.37 g/L,分配比D=0.45,酸化料的乳酸浓度控制在25%左右;萃取剂相比2.5;10级逆流萃取,6级逆流反萃;经脱油、脱色、水洗,浓缩等工序得到合格乳酸产品,用萃取与反萃技术从复杂体系中提取乳酸是可行的。
猜你喜欢
农产品加工(2022年9期)2022-06-17
新乡医学院学报(2022年2期)2022-03-22
中国油脂(2022年1期)2022-02-12
中国果业信息(2021年10期)2021-12-07
农业研究与应用(2021年3期)2021-08-23
粮食与食品工业(2021年4期)2021-08-19
农技服务(2021年3期)2021-06-16
广东农业科学(2020年9期)2020-11-10
燃料化学学报(2020年3期)2020-05-07
食品界(2019年8期)2019-10-07
