
均相催化CO2/H2还原羰基化合成高值化学品研究进展
2021-06-02 11:38张雪华曹彦伟陈琼遥沈超仁何林
物理化学学报 2021年5期
张雪华,曹彦伟,陈琼遥,沈超仁,何林,3,*
1盐城师范学院化学与环境工程学院,江苏 盐城 224007
2中国科学院兰州化学物理研究所,羰基合成与选择性氧化重点实验室,兰州 730000
3中国科学院洁净能源创新研究院,辽宁 大连 116023
1 引言
CO2是构成温室气体的主要成分,实现其高效利用不仅可有效解决环境问题,还可创造经济价值1。依照巴黎协定在联合国气候变化公约框架下所制定的目标,控制CO2在大气中的浓度,将全球平均气温升幅控制在1.5 °C之内2。随着清洁能源的普及和CO2捕集技术的成熟,通过化学固定的方式将廉价易得、环境友好的CO2作为可再生C1资源转化成高值化学品,符合绿色与可持续发展的要求3-5。
目前,全世界范围内CO2资源化的年利用量已达到1亿吨,主要是通过非还原性转化与氨、氯化钠、苯酚钠和环氧化物等反应生产尿素、纯碱、水杨酸和碳酸酯类产品,以及作为超临界溶剂使用6-10。拓宽基于CO2还原性转化的化学品合成新路线是开发其资源化利用的热点。其中,通过热化学的催化方法,将惰性CO2还原,进而转化为甲醇、甲酸、烯烃、烷烃等能源产品相关研究不断创新,正在向产业化迈进,譬如CO2加氢制甲醇技术已经进入工业化放大阶段11-13。如能以清洁、高原子经济性的H2作为还原剂实现惰性CO2还原性转化,结合羰基化过程构筑C―O、C―N和C―C键,合成醛/醇、羧酸、酯、酰胺等化学品,将极大扩展由CO2高值化利用的范围与种类,无论在资源利用性和环保性等方面都表现出极具潜力的应用前景14-16。
其中均相催化CO2还原羰基化制备高值化学品是CO2化学转化利用研究的前沿领域之一,但实际反应中仍面临着众多的挑战。首先,CO2因其碳原子处于最高氧化态+4价,导致在反应的过程中金属催化剂活性物种对CO2的C=O加成能垒过高,通常需要高温高压苛刻的反应条件以及贵金属催化剂才能实现其活化。同时,均相催化CO2、H2参与的还原羰基化反应多是串级反应,一般都涉及多个竞争反应,导致目标产物的选择性并不理想。此外,受底物的位阻效应和电子效应的影响,底物的适用性范围有一定的局限性。随着新型表征技术和理论计算的发展,在揭示反应机理方面及CO2中碳―氧键活化和转化也取得一些进展。因此,结合现代表征手段发展更加高效的催化体系、非贵金属体系甚至无金属催化剂体系,实现以CO2为羰源、H2为还原剂在相对温和的条件下制备高值化学品。
近年来,均相催化CO2还原方面的研究成果大量涌出,相关的综述论文也在不断的更新11,13,15,17-21。本综述以CO2为羰基化试剂以及相对于氢硅烷或氢硼烷等更加清洁的H2为还原剂,通过金属催化剂均相催化制备高值化学品。主要从CO2与烯烃羰基化、胺羰基化、醇/醚羰基化以及其它羰基化反应进行阐述。希望通过本文的总结,为系统的认识和发展均相催化CO2还原羰基化反应提供一定的参考和借鉴。
2 H2作为还原剂用于均相催化CO2还原羰基化合成化学品
2.1 烯烃羰基化反应
烯烃羰基化的产物在精细和大宗化学品制造中皆具有重要价值,该过程通过C1资源定向增碳转化烯烃22,23。工业上主要采用有毒、易燃CO作为羰源24-27,如能利用无毒、易得CO2代替CO作为羰源,对开发绿色、高效的烯烃羰基化催化合成方法具有重要意义。以H2作为还原剂、CO2为羰源的烯烃羰基化过程可看作是CO2还原和烯烃羰基化的耦合,具体而言,首先通过逆水煤气变换(RWGS,CO2+ H2→ CO + H2O)将CO2还原成活泼的CO,随后CO与烯烃羰基化反应生成目标产物。
CO2和烯烃羰基化过程首先是吸热的逆水煤气变换。在工业过程中RWGS主要用于调节费托合成使用的合成气的碳/氢比。常用Cu-Ni、Cu-Zn/Al2O3等多相催化剂28,且反应温度大多超过250 °C,与后续的CO和烯烃的羰基化过程反应需要低温不匹配。均相催化RWGS过程的温度则低得多,常用均相催化剂主要是Rh和Ru络合物。均相催化RWGS要求M-H加成到CO2的C=O上得到形成M-COOH络合物中间体,或者金属的富电子中心进攻CO2中具有弱亲电性的碳原子形成MCOO-中间体,然后脱去羟基形成羰基配体进而释放一分子CO。Ford等最早发现Ru3(CO)12具有一定的逆水煤气变换的能力29。在1993年,Tominaga等发现Ru3(CO)12催化剂在碘化物为助剂条件下,催化CO2和H2还原成甲烷,反应过程中检测到CO和甲醇30。碘化物的加入有效的抑制金属钌的沉积,提高反应的活性。单核过渡金属络合物也具有催化RWGS的活性31,Koinuma首先报道了RhCl(PPh3)3在一定压力的CO2/H2下催化RWGS过程32。在1977年Eisenberg等发展了[RhI2(CO)2]-/H+/I-体系在低于95 °C的条件下实现RWGS33。在1995年,Tominaga等发现在环氧乙烷存在情况下RuCl2(PPh3)2络合物可以间接催化RWGS过程34。后续研究者又报道了单核钌络合物[PPN][RuCl3(CO)3]催化RWGS35,在反应温度180 °C时的TON高达96。把该催化剂固载在离子液上,催化体系具有优异的RWGS活性,并能重复使用36。
在2000年,Tominaga等第一次报道了用Ru络合物催化CO2、H2和烯烃的氢甲酰化反应,主要产物为醇37。该反应过程首先是CO2通过RWGS生成CO,进而CO与烯烃羰基化生成醛,最后醛被H2还原得到醇(图1)。研究者使用H4Ru4(CO)12为催化剂及LiCl为助剂转化环己烯得到收率为88%环己基甲醇。后续又对CO2和烯烃的氢甲酰化过程进行更加细致的研究38,发现不同的卤原子对活性有很大的影响(I-< Br-< Cl-),正好和质子亲核能力相对应。同时借助ESI-MS分析发现四种活性物种:[HRu3(CO)11]-、[H3Ru4(CO)12]-、[RuCl3(CO)3]-和[RuCl2(CO)3(C6H10)]-,结合对照实验提出可能的机理。研究者通过多核钌簇和离子液体构建可重复使用的催化体系,对CO2和末端烯烃氢甲酰化有很好活性和直链醇的选择性39。
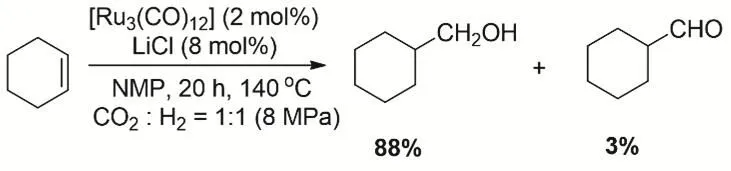
图1 Ru催化CO2、H2和环己烯氢甲酰化到醇Fig. 1 Ruthenium-catalyzed hydroformylation of cyclohexene with CO2 and H2.
Hakka等在2003年和2009年相继报道了Ru络合物催化CO2、H2和1-己烯的还原氢甲酰化生成醇的反应40,41。H4Ru4(CO)12、Ru3(CO)12、[Ru(CO)3Cl2]2以及[Ru(CO)4]n都具有一定的CO2和烯烃的氢甲酰化活性;通过对照实验论证了卤素是产生活性中心必不可少的要素;ESI-MS表明[Ru(CO)4]n在卤素阴离子存在下原位转化为具有RWGS能力和烯烃氢甲酰化能力的活性物种,譬如[Ru4(CO)12]4-、[HRu3(CO)11]-和[Ru(CO)3Cl3]-。在2014年,Beller等首次把亚膦酸酯配体配位修饰Ru3(CO)12应用到CO2、H2与烯烃氢甲酰化生成醇的反应42。膦配体的加入或修饰有效的抑制烯烃的加氢反应,提高的烯烃羰基化生成醇的收率。通过一系列的控制实验发现配体的加入,主要是促进了氢甲酰化过程,没有加速RWGS过程(图2)。Dupont等发现咪唑鎓盐1-丁基-3-甲基咪唑氯化物([Bmim]Cl)或3-丁基-1,2-二甲基咪唑氯化鎓盐([Bmmim]Cl)和Ru3(CO)12形成氮杂环卡宾-钌络合物,实现逆水煤气变换-氢甲酰化-羰基还原的耦合,将烯烃转化为增加一个亚甲基的醇,同时发现体系适量的磷酸有利于反应进行43。

图2 膦配体应用于Ru催化CO2、H2和烯烃的氢甲酰化过程Fig. 2 Increased selectivity and substrate scope for the ruthenium-catalyzed hydroformylation of olefins with a bulky phosphite ligand.
丁奎岭等报道了Rh(acac)(CO)2和螺环骨架的亚膦酸酯配体构成的催化体系催化CO2、H2、聚甲基氢硅氧烷(PMHS)和烯烃的氢甲酰化到醛的反应(图3)。反应的TON在12 h后高达1000000,同时能得到很好的直链醛的选择性。反应的过程中没有检测到CO的生成44。
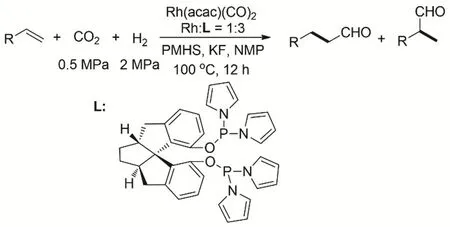
图3 Rh催化烯烃与CO2/H2的氢甲酰化反应Fig. 3 Rhodium-catalyzed CO2/H2 and olefins hydroformylation.
我们课题组利用由兼具逆水煤气变换和羰基还原催化能力的[Ru(CO)3]Cl3]-以及氢甲酰化催化剂Co2(CO)8构成的双催化剂体系实现以CO2为羰源的烯烃氢甲酰化合成醇45。在研究中,发现了向体系中加入适当酸性的有机酸,能在不影响氢气活化的前提下显著加速逆水煤气变换中CO的生成速率,进而推动后续氢甲酰化和羰基还原反应的发生,并有效抑制烯烃直接氢化。该催化体系能够将柠檬烯生物质平台分子转化为增加一个碳的相应醇类分子,还能将碳四馏分中的廉价副产物二异丁烯转化为高附价值的异壬醇,展示了该催化方法在生物质利用和廉价原料高值化转化方面的潜力。理论计算结果揭示酸参与和无酸的逆水煤气变换过程,酸参与时Ru-COOH的质子化脱羟基生成CO过程无显著能垒(图4)。
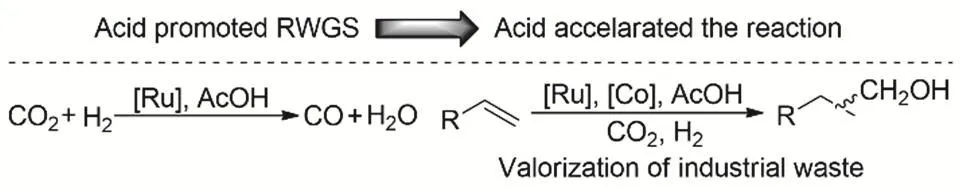
图4 酸促进RWGS进而加速CO2和烯烃氢甲酰化反应Fig. 4 Acid promoted RWGS thereby accelerating hydroformylative with CO2.
烯烃氢甲胺化是逆水煤气变换-氢甲酰化-醛与胺的耦合-烯胺或亚胺的还原过程。反应中需要抑制烯烃直接加氢同时避免胺和CO2/H2的N-甲酰化或N-甲基化,甚至是胺和CO2直接形成脲或碳酸铵盐,反应过程复杂且具有挑战性,对反应的化学选择性要求很高。在2009年,Eilbracht等报道了Ru3(CO)12、LiCl、苄基三甲基氯化铵(BTAC)构建的催化体系催化CO2、H2、吗啉和烯烃通过氢甲胺化生成吗啉氮原子上烷基取代的叔胺46。能够得到烯烃氢甲胺化产物的收率为35%-98% (图5)。Dupont等在2016年使用Ru3(CO)12/咪唑鎓盐氯化物催化体系催化CO2、H2、烯烃和伯胺或叔胺的氢甲胺化47。该催化体系使反应能在更低的温度下实现。整个反应过程首先是逆水煤气变换,进而CO与烯烃氢甲酰化生成醛,醛与胺耦合,最后亚胺或烯胺被H2还原。

图5 Ru催化CO2、H2和烯烃氢胺甲基化Fig. 5 Ruthenium-catalyzed hydroaminomethylation of alkenes with CO2 and H2.
羧酸合成的经典方法是醇或烯烃的氧化或羰基化。利用CO2作为C1原料在还原剂作用下与烯烃还原羧化,通过构建C―C键制备羧酸是具有挑战的新路线48。在2013年,Leitner等发现由[RhCl(CO)2]2/PPh3构成的催化剂体系将CO2、H2和烯烃氢羧基化合成增一个碳的羧酸49。通过控制实验和同位素实验推测游离态的甲酸中间体在该条件下不太可能会形成50,反应首先是逆水煤气过程生成CO,接着CO和Rh的络合物配位并发生烷基的迁移插入,最后Rh-酰基物种发生水解生成羧酸(图6)。

图6 催化CO2、H2和烯烃氢羧化合成羧酸Fig. 6 Catalytic synthesis of carboxylic acids from olefins and CO2/H2.
醇是廉价、易得的非气态氢给体型还原剂。空气中稳定且对水、氧不敏感的液态还原剂甲醇用作氢转移还原CO2具有操作上的优势。2014年,Beller等报道了Ru3(CO)12/[Bmim]Cl催化体系催化CO2、甲醇和烯烃的氢酯化反应(图7)。第一次提出了CO2作为羰源的一个新过程,利用醇的还原性,原位还原CO2生成CO用于接下来的羰基化反应,最后再与醇生成酯。醇在反应中是还原剂、原料以及溶剂。通过控制实验和同位素实验证实了产物中的羰基来自于CO2,并提出可能的反应路径。各种烯烃和醇均可顺利反应得到相应的羧酸酯51。

图7 CO2、醇和烯烃氢酯化Fig. 7 Alkoxycarbonylation of olefins with CO2 and alcohols.
我们课题组2018年报道了[Ru(CO)3Cl2]2-Co2(CO)8双金属体系应用于CO2与烯烃的氢酯化反应中52。在同等活性的条件下,他们使用了更少的Ru催化剂和离子液,整个体系重复使用五次活性没有明显下降(图8)。通过一系列的控制实验和同位素实验论证,发现Ru催化剂主要负责催化CO2的逆水煤气变换的过程,而CO和烯烃的羰基化过程主要依靠Co催化剂。
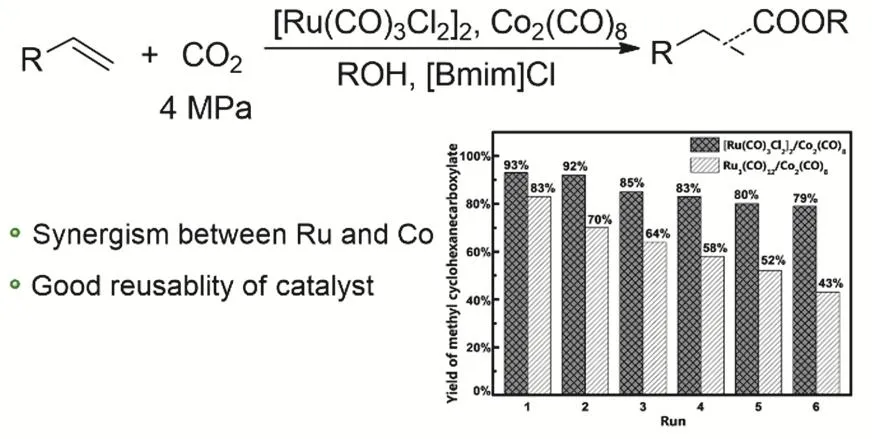
图8 Ru-Co双金属催化体系用于CO2和烯烃氢酯化Fig. 8 Alkoxycarbonylation of olefins with CO2 and alcohols by Ru-Co bimetallic catalytic system.
2.2 胺羰基化/胺甲基化反应
酰胺化合物是具有广泛用途的重要化学品,大量存在于精细化学品,天然产物和聚合物中。在工业上主要通过有机胺与CO羰基化反应路线制备。随着可持续发展与绿色化学合成的要求,利用易得、无毒的CO2代替CO合成酰胺化合物越来越受研究者的关注。与CO合成酰胺化合物不同的是CO2作为羰源时需要在还原剂的作用下,通过三组分才能实现胺的羰基化。
胺的羰基化反应主要是N-甲酰化反应。早在1970年代,Haynes等首次报道通过均相催化CO2、H2和二甲胺羰基化合成N,N-二甲基甲酰胺(DMF)(图9)。很多金属催化剂对该反应都有很高效的催化活性,譬如CoH(dppe)2、(PPh3)2(CO)IrCl和CuCl(PPh3)3,其中(PPh3)2(CO)IrCl活性最高53,反应的TON可以达到1200。随后贵金属Pd54、Pt55,56、Ru57,58的络合物也被用于该反应的研究,可大大降低反应的条件,并显示良好的催化性能。例如,在超临界反应条件下(130 °C,CO2为13 MPa,H2为8 MPa),RuCl2[P(CH3)3]4催化合成DMF的TON可以达到370000,TOF也有10000 h-1。主要由于反应处于超临界条件,整个反应在均相状态,提高反应速度57。Baiker等报道的带有双齿磷配体的Ru络合物RuCl2(dppe)2是迄今为止最高效的合成DMF的催化剂59。在反应条件100 °C、CO2压力为13 MPa和H2压力为8.5 MPa时,反应的TON高达740000。2010年,Beller等第一次报道铁络合物催化二甲基胺与CO2合成DMF。Fe(BF4)2·6H2O作为催化剂前体和P(CH2CH2PPh2)3作为配体60得到DMF的收率为75%以及反应的TON为727。随后他们又发展更加高效的铁催化剂以三(2-(二苯基膦基)苯基)膦为配体61,反应的TON达到5104。同时他们还发现Co(BF4)2·6H2O和P(CH2CH2PPh2)3也能有效的催化二甲基胺与CO2、H2合成DMF62,反应的TON为1308。
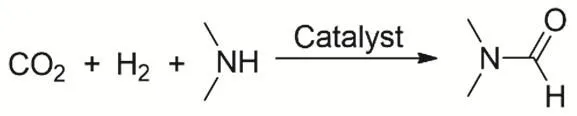
图9 CO2、H2与二甲胺合成DMFFig. 9 Catalytic synthesis of DMF from dimethylamine with CO2 and H2.
为了拓展CO2、H2参与的有机胺N-甲酰化的适用性范围,研究者发展一系列高效的催化体系。2015年,丁奎岭课题组报道Ru pincer络合物能够高效催化有机伯胺或仲胺的N-甲酰化反应(图10),反应的TON最高可达1940000。在最优的条件下(Ru催化剂0.01% (摩尔分数),CO2与H2的压力均为3.5 MPa,四氢呋喃为溶剂,反应温度120 °C),可以得到52%-99%相应的甲酰化产物的收率。使用该类催化剂制备DMF,催化剂可回收利用,重复使用12次之后催化活性基本保持不变63。
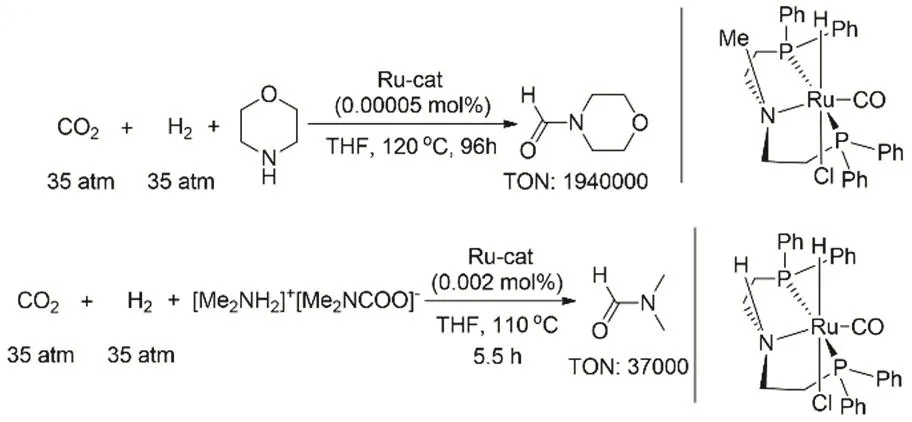
图10 Ru pincer催化CO2、H2与胺的甲酰化反应Fig 10 Ru pincer catalyze formylation of amine with CO2 and H2.
芳香胺由于碱性较弱,一般CO2、H2参与的芳香胺N-甲酰化反应的活性通常较低。Jessop等发现碱的加入可以大大提高反应的效率(图11),在采用RuCl2[P(Me3)3]4催化剂时,额外加入2当量的有机碱DBU,在反应温度100 °C条件下,可以转化苯胺到甲酰基苯胺的收率达到85%64。
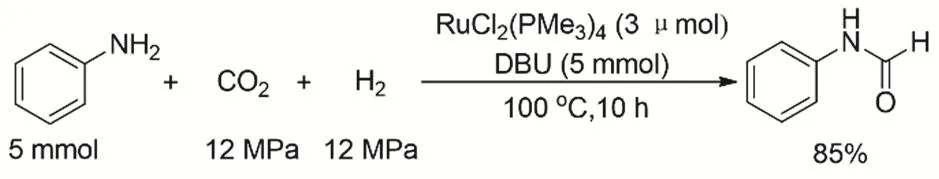
图11 CO2、H2与芳香胺的甲酰化反应Fig. 11 Formylation of aromatic amines with CO2 and H2.
非贵金属络合物也可用于CO2、H2与有机胺的N-甲酰化反应65。Co pincer络合物结合tBuOK,在反应条件下(Co 5%,NaHBEt35%,tBuOK 5%,(均为摩尔分数),CO2和H2的压力均为3 MPa,甲苯为溶剂,反应温度150 °C),能够催化一系列脂肪胺和仲胺的N-甲酰化反应,收率为60%-90%。反应机理推测(图12),Co pincer首先与tBuOK反应生成不饱和的中间体;在H2存在时产生一价的Co氢负;接着CO2插入Co-H键;由于过量胺存在,通过与pincer配体去质子化,形成甲酸盐;甲酸盐脱水,形成甲酰胺。
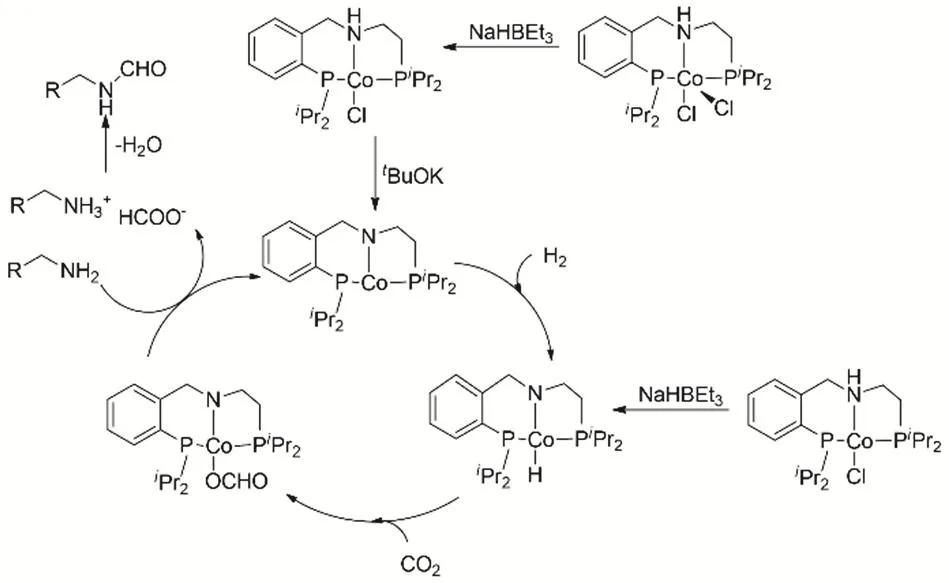
图12 Co pincer催化CO2、H2与胺的甲酰化反应Fig. 12 Co pincer catalyze formylation of amine with CO2 and H2.
当胺类化合物含有不饱和基团与CO2、H2发生N-甲酰化反应时,一般都面临不饱和基团的加氢反应,结果导致反应的选择性差。Cu(OAc)2/4-二甲基氨基吡啶(DMAP)催化体系可以实现含不饱和基团的胺类化合物的N-甲酰化反应(图13)。在最优的反应条件下(Cu催化剂为10% (摩尔分数),DMAP为2 mmol,四氢呋喃为溶剂,CO2和H2压力均为4 MPa,反应温度90 °C),得到甲酰化产物的收率31%-95%,同时底物中的碳碳双键、碳氮双键、羰基和酯基能够很好的兼容66。
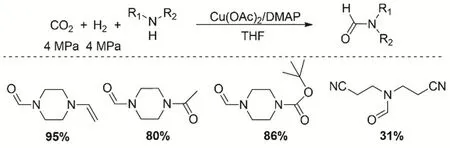
图13 CO2、H2与不饱和胺的甲酰化反应Fig. 13 Formylation of unsaturated amine with CO2 and H2.
刘志敏等报道一条新颖的合成不对称N,N-二取代甲酰胺路径。通过CoF2、P(CH2CH2PPh2)3和K2CO3催化体系催化还原耦合伯胺、醛以及CO2/H2制备(图14)。反应机理考察表明伯胺与醛耦合形成亚胺,亚胺还原生成仲胺,仲胺与CO2加氢的产物HCOOH反应最终形成不对成N,N-二取代甲酰胺67。
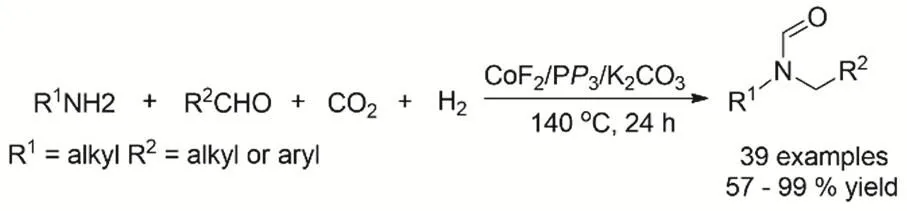
图14 CO2、H2参与合成不对称N,N-二取代甲酰胺Fig. 14 Catalytic synthesis of asymmetric N,N-disubstituted formamide by using CO2 and H2.
常见的非贵金属Mn和Fe也具有催化CO2/H2和胺的N-甲酰化反应(图15),Khusnutdinova等第一次报道了具有生物活性和强配体的Mn络合物可以有效的催化CO2加氢与仲胺发生甲酰化反应68。Bernskoetter等报道带有PNP配体的二价Fe羰基螯合物能够高效的转化多种仲胺底物和CO2/H2生成甲酰化产物69,产物的收率高达92%,反应的TON也有8900。

图15 Mn和Fe络合物催化CO2、H2与胺甲酰化反应Fig. 15 Mn and Fe complexes catalyze the formylation of amines.
有机胺N-甲酰化产物上的羰基进一步还原可以转化成-CH3,甲基胺是许多具有生物活性化合物中的关键结构基序,因此是众多药物和农药化学品的重要组成部分70。在N官能团上引入甲基一般要涉及有危险的甲基化试剂,像碘甲烷、硫酸二甲酯等。另外碳酸二甲酯、甲醛和甲酸也可以作为C1源用于胺甲基化试剂(Eschweiler-Clark 甲基化)71。尽管如此,CO2才是被认为是胺甲基化的最清洁的C1源。最早CO2和H2用于N-甲基化反应的是多相催化剂(图16),Baiker等报道以Cu/Al2O3为催化剂,CO2、H2和NH3为原料气,在固定床微反应器上合成甲胺以及二甲胺、三甲胺72。在此基础上,研究者又发展了Ni-、Pt-、Co-、Fe-、Pd-、Au-基等催化剂用于CO2和H2参与的N-甲基化反应,可以高选择性地得到单甲基化的胺73。但反应体系还是存在反应速率较低,且当反应温度过高时副产物CO、CH4明显增多。

图16 多相催化剂CuAlOx用于CO2、H2和NH3的N-甲基化反应Fig. 16 N-methylation of NH3 with CO2 and H2 by heterogeneous catalyst CuAlOx.
均相催化CO2和胺的N-甲基化反应最初使用的是硅烷/硼烷作为还原剂。直到2013年,Klankermayer/Leitner等在CO2和H2的氛围下,第一次实现了伯胺和仲胺的甲基化74。三乙烷基Ru基催化剂是该体系成功构建N-甲基化的关键,Ru基催化剂的选择主要受CO2还原合成甲醇的催化剂启发75。另外,易合成的络合物[Ru(triphos)(tmm)](tmm = 三甲叉基甲烷)在CO2和H2的氛围下可以高效的实现芳胺的甲基化(图17)。通过5% (摩尔分数)的酸和非配位的阴离子预活化催化剂使催化剂具有最好的活性。在CO2/H2(2/6 MPa)、反应温度140-150 °C的四氢呋喃(THF)溶液中,可以转化大部分的芳基伯胺和芳基仲胺为二甲胺和甲胺,甲基化的收率> 90%。但是对脂肪类的胺的活性却很低。
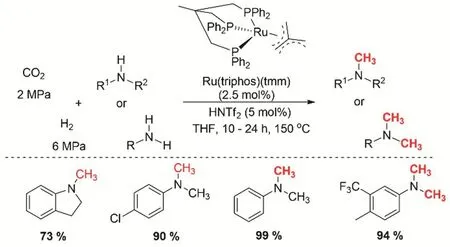
图17 Ru催化CO2、H2与胺的甲基化反应Fig. 17 Ruthenium-catalyzed methylation of amines with CO2 and H2.
随后,Beller等报道了类似的路线用于芳香胺和脂肪胺的N-甲基化。在CO2/H2存在的条件下,通过[Ru(acac)3]/triphos/H+原位形成络合物作为催化活性组分,同时在体系添加LiCl可以提高脂肪胺N-甲基化的活性和选择性(图18)。LiCl的加入可能是与THF的氢原子作用提高了酸的质子化能力76。通过一系列的控制实验表明甲基化的胺首先形成了甲酰胺中间体,接着被还原成甲基胺。反应的过程中虽然检测到了甲醇的产物,但是甲醇作为甲基化的试剂的产物基本是忽略不计的。
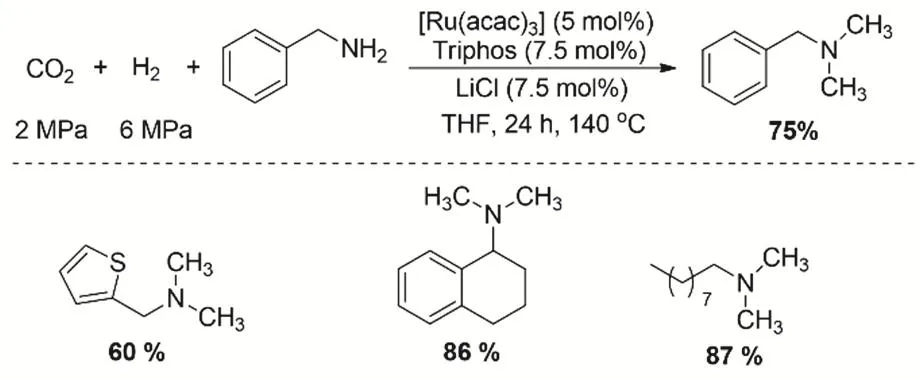
图18 在存在LiCl助剂的条件下Ru催化CO2、H2与脂肪胺/芳香胺的甲基化反应Fig. 18 Ruthenium-catalyzed methylation of amines with CO2 and H2 in the presence of LiCl as additive.
Leitner等报道[Ru(triphos)(tmm)]/三氟甲基磺酸盐在CO2/H2的气氛下实现氨气和氯化铵的三甲基化(图19)。在有机溶剂中合成三乙胺的过程Lewis/Brønsted酸是必需的。在氯化铵三乙基化的过程中水和有机溶剂的定量比是至关重要的,同时水/有机两相有利于产物的分离77。

图19 CO2、H2与NH3/NH4Cl合成三乙胺或其盐酸盐Fig. 19 Synthesis of trimethylamine TMA and its hydrochloride starting from NH3 or NH4Cl with CO2 and H2.
尽管早在上世纪90年代就有报道CO2/H2参与的胺甲基化反应,直到近年才实现贵金属和非贵金属催化剂的在温和条件下催化甲基化产物的高效合成,但仍然存在催化剂活性和选择性较低的问题。引入碱性基团或缺电子催化中心有效活化CO2分子,以及增强N-亲核试剂的亲核性,都有利于C―H键的形成。胺甲基化过程经常形成甲酰胺中间体,因此在催化剂和配体设计的时候可借鉴酰胺还原催化剂的思路有望获得更高活性的催化剂。
氮杂环化合物及其衍生物是一类重要有机化学品,广泛用于有机物和药物中间体。其中苯并杂环化合物可通过邻苯二胺与羧酸或其衍生物、酯、酰胺、内酯、腈和醛等在高温下环化而合成。由CO2、H2和胺羰基化构建C―N键,是符合绿色化学发展的。刘志敏等报道采用RuCl2(dppe)2为催化剂,通过CO2、H2和邻苯二胺反应合成苯并咪唑化合物78。反应在超临界条件下CO2和H2的总压为15 MPa、温度120 °C时,含有吸电子基团和给电子基团的邻苯二胺底物生成苯并咪唑的收率均能达到> 90%。且反应在无溶剂条件下进行,反应过程中只有副产物水生成,符合绿色反应的路线。后续工作中研究者进一步发展非贵金属催化体系CoF2/PP3/CsF (图20),通过胺邻位取代与CO2和H2还原环化制备氮杂环化合物79。
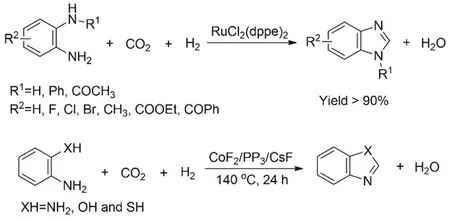
图20 Ru和Co络合物催化CO2、H2与胺的还原环化Fig. 20 Ru and Co complexes catalyze the reductive cyclization of amines with CO2 and H2.
2.3 醇/醚羰基化反应
乙醇是重要的化工原料且用途广泛,工业上主要通过发酵法、乙烯水合法、煤化工法等制备,由CO2和醇/醚羰基化合成乙醇意义非凡(图21)。2018年,韩布兴等使用了Ru-Co双金属体系,在CO2和H2的条件下,用二甲醚合成乙醇。整个反应的转化率达到71.1%,乙醇的选择性可以高达94.1%,催化剂连续套用5次活性没有明显下降,同时乙醇的收率高于以前报道过的CO2/CO合成乙醇的路线的收率80。他们通过同位素实验验证了产物乙醇的烷基来自于二甲醚(DME),羰基来自于CO2,但是产物中的H随H2压力的增大才会有更多的氢来自于氢气。该反应的机理大致是,首先是Li+促进DME的CH3―O键的弱化,在Lewis酸的作用下Co的插入形成CH3Co*,然后Ru催化的逆水煤气变换原位生成的CO与CH3Co*结合形成CH3COCo*,最后金属键断裂H2还原乙醛生成目标产物乙醇,一部分的CH3O―形成甲醇。接着研究者又在更加温和的条件下,发展[RuCl2(CO)3]2/Co4(CO)12双金属催化体系用于CO2、H2和甲醇羰基化制备乙醇81。反应体系中LiI是促进剂,N-乙基-2-吡咯烷酮(NEP)作为溶剂,反应温度为160 °C时,生成乙醇的Ru的TOF高达7.5 h-1,同时乙醇的选择性为65%。反应首先发生RWGS,接着CO、H2与甲醇羰基化生成乙醇。接着研究者又发展更加温和的单金属催化剂Ru3(CO)12/[Bmim]Cl体系,在120 °C的反应条件下CO2、H2和甲醇羰基化合成乙醇82。同时研究者还报道CO2、H2与多聚甲醛羰基化制备乙醇83。
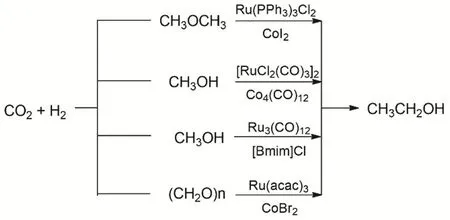
图21 CO2、H2与二甲醚/甲醇/多聚甲醛羰基化制备乙醇Fig. 21 Catalytic synthesis of ethanol from dimethyl ether/methanol/paraformaldehyde with CO2 and H2.
羧酸是重要的大众化学品,目前通过CO2和醇/醚羰基化制备是十分具有挑战的课题。主要受热力学的限制,需要强还原剂和苛刻的反应条件。2016年,韩布兴等报道了以CO2作为羰基化试剂、H2作为温和还原剂,通过甲醇还原羰基化制备乙酸的新路线84。Ru3(CO)12/Rh2(OAc)4催化体系是实现乙酸高收率的关键。在CO2和H2存在的条件下,以咪唑为配体、LiI为促进剂,DMI作为溶剂,反应温度200 °C,使甲醇羧酸化合成醋酸,TOF达到30.8 h-1,催化剂连续套用5次活性保持不变(图22)。通过CO2羰基化合成的醋酸,这是醋酸合成的一个全新路线,对整个合成化学有很重要的影响。通过控制实验和同位素实验认为反应的步骤是:1) 甲醇在Li+的促进作用下原位转化为CH3I,主要由于在高温下Lewis酸阳离子Li+的促进形成比较活泼的活性中间体CH3I85。2) 通过NMR对13CH3OH同位素实验的表征可以观察到中间体CH3Rh*I形成。3) CO2插入到CH3Rh*I中形成CH3COORh*I,整个过程中Rh催化剂起主要的作用。由于咪唑可以增强Rh原子的电子云密度,有利于CO2的插入86。4) 下面的步骤主要是CH3COORh*I的还原和醋酸形成的过程,同位素实验和控制实验表明该步骤的主要活性中心是Ru。气体的主要产物为CH4,未检出到CO。在此工作基础上,研究者又发展了更加简单和温和的Rh2(CO)4Cl2/4-甲基咪唑/LiI催化体系应用于CO2/H2和甲醇反应,在反应温度为150 °C时,反应中就检测到了乙酸87;反应到180 °C是乙酸的产率达到81.8%,反应的TOF达到26.2 h-1。
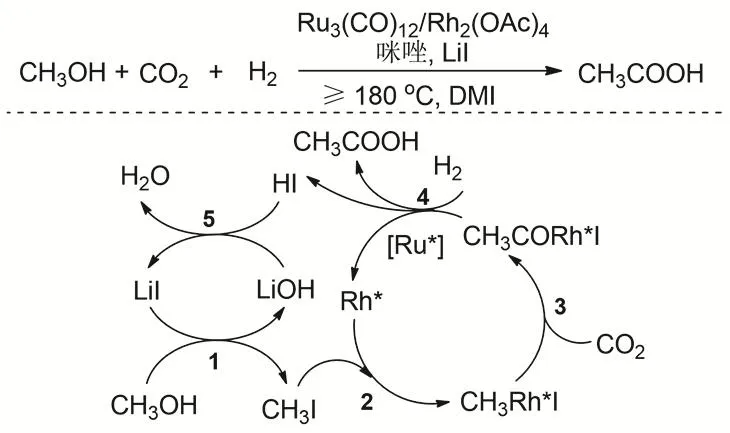
图22 CO2、H2与甲醇羰基化制备乙酸Fig. 22 Synthesis of acetic acid by methanol carbonylation with CO2 and H2.
研究者进一步以醚类化合物、CO2和H2为原料,通过构建IrI4/LiI高效催化体系,成功制备了长链羧酸。在170 °C温度条件下,在乙酸溶剂中该反应可高效进行,各种醚均可转化为对应的高级羧酸(图23)。机理研究表明,底物醚首先在催化剂作用下转化为烯烃,烯烃进一步转化为烷基碘化物;该类碘化物再与经逆水煤气反应原位生成的CO反应,生成高级羧酸。该研究成果为CO2转化和高级羧酸的合成提供了一个新策略88,89。
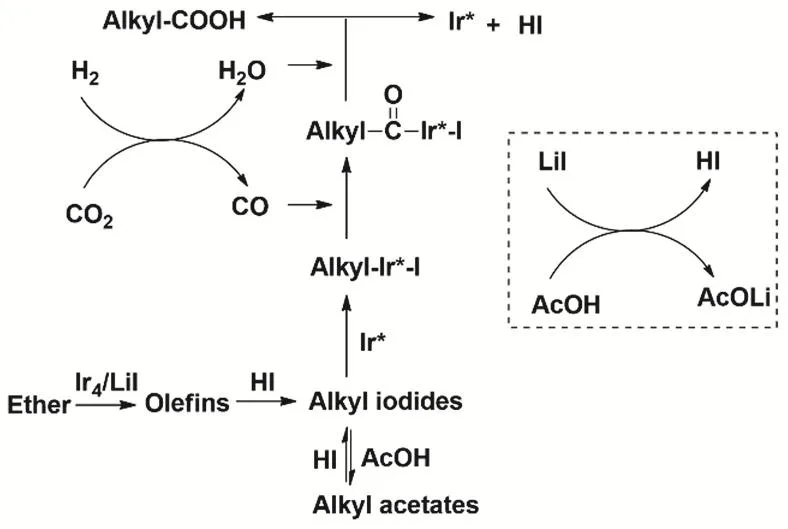
图23 CO2、H2与醚羰基化制备高级羧酸Fig. 23 Synthesis of higher carboxylic acids from ethers, CO2 and H2.
CO2、H2合成C2+的产物往往都涉及C―C偶联和C―OH的形成,同时生成产物具有重要的意义。譬如由CO2加氢合成乙醇和长链醇(C2+醇),一般都要经历低碳醇羰基化到高碳醇的过程。但是,受制于选择性问题,CO2/H2合成C2+醇一般都需要特别的催化体系和更为苛刻的反应条件。迄今报道主要集中在多相催化剂上,相对于负载型单金属催化剂,具有协同效应的杂金属催化剂(CuFe、CuZnFe、CoMo等)90能够更加高效的转化CO2和H2到C2+OH。CoMoS在340 °C可以催化CO2氢化合成醇,C2+OH的产率最高为35.6%91。以CuZnFe/K为催化剂在反应温度300 °C得到C2+OH的选择性达到87.1%92。相对而言,均相催化体系能在相对温和的条件下,实现催化C2氢化到C2+OH。但是目标醇的高选择性仍然是挑战性的课题。
在1994年,Tominaga等就发现Ru3(CO)12和Co2(CO)8双金属催化体系在KI存在的情况下均相催化CO2还原到乙醇31。一定比例的Co/(Co + Ru)催化剂在整个的反应过程中没有金属析出,当该比例大于0.5时催化剂反应后有金属析出,所以当该比例小于0.5时他们认为在整个的反应过程中Co取代一部分的Ru形成了一个比较稳定的簇。而Fe或Mo就没这种双金属协同催化的作用。当一定量Co取代一定量的Ru时反应活性没有下降,乙醇的收率还有轻微的提高,由此得到Ru主要的催化作用是把CO2还原成CO,而CO到乙醇主要是Co的催化剂起作用。但是产物中C2+的产物醇的选择性相比于C1的产物CH3OH和CH4还是相对比较低。关于如何在温和条件下高选择性催化该反应依然是很具有挑战的课题。直到20年后,韩布兴等采用[Ru3(CO)12]/[Rh2(CO)4Cl2]双金属催化体系以KI为促进剂93,在反应条件(CO2和H2均为4 MPa,DMI为溶剂,200 °C,12 h)可以得到C2+产物的收率高达96.4%(图24)。LiI在反应中起到了重要的作用,Li+有较强的Lewis酸性,起到稳定催化剂的作用;而且I-的亲核性能够促进反应链的增长。Ru-Rh双金属的协同催化能力通过控制实验被证明。
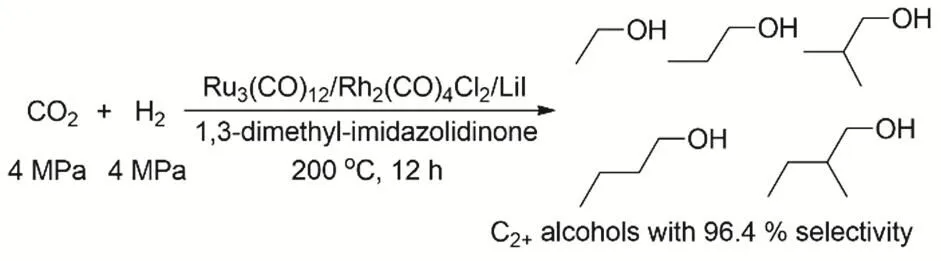
图24 RuRh双金属体系催化CO2和H2合成C2+OHFig. 24 Synthesis of C2+ alcohols by CO2 hydrogenation with a homogeneous bimetallic Ru-Rh system.
接着研究者又发展了Ru3(CO)12/Co4(CO)12双金属催化体系94,在共催化剂[PPN]Cl和促进剂LiBr共同作用下,可以高效的催化CO2/H2到C2+OH(图25)。同时该双金属催化体系能够重复使用5次依然保持很好的活性。Ru-Co双金属之间存在明显的协同作用,[PPN]Cl和LiBr的加入提高了催化活性和选择性。反应机理推测Ru催化CO2还原生成CH3OH和CO。CH3OH原位与CO还原羰基化转化成乙醇。丙醇的形成与形成乙醇的类似。少量的异丁醇是甲醇和丙醇发生Guerbet反应形成的95。在反应过程中,Ru催化剂催化CO2加氢生成CH3OH和CO,并在Br-促进下催化甲醇羰基化到乙醛,同时是乙醛氢化到乙醇;Co催化剂的主要作用加速甲醇的还原羰基化到乙醇和乙醛以及C2+醇的生成96。

图25 Ru-Co双金属催化体系催化CO2和H2合成C2+OHFig. 25 Synthesis of C2+ alcohols by CO2 hydrogenation with a homogeneous bimetallic Ru-Co system.
2.4 其它羰基化反应(卤化物羰基化、甲基化等)
烷基卤化物中与卤素相连的碳原子具有亲电性,可以发生偶联反应。以CO2作为羰源、H2作还原剂与卤化物反应合成羧酸产品是一条有效的途径。在1995年,Fukuoka等使用Ru/Co或Ni/Co双金属催化剂在CO2、H2的气氛下均相催化有机碘化物羧基化97。该反应的显著特征是使用双金属催化剂协同催化生产羧酸化合物(图26)。反应物选择是CH3I,在单独使用Ru3(CO)12或Co2(CO)8时反应体系中没有检测到醋酸生成,当使用Ru3(CO)12/Co2(CO)8双金属催化剂时生产醋酸的TON为17(以Ru原子)。在反应的过程中也检测到副产物CH4和CO,反应TON分别为50和58。尽管推测是CO2直接插入M-CH3的机理,但是也没排除通过RWGS反应原位生成的CO进行羰基化的可能性。

图26 CO2、H2与CH3I羰基化制备乙酸Fig. 26 Synthesis of acetic acid from methyl iodide with carbonylation CO2 and H2.
药物研究中神奇甲基效应与C―H键的甲基化有很大的关系。通过CO2直接构建C―C键,主要受限C―H的羰基化的难度,所以相关报道不多98。Beller课题组第一次报道Ru络合物催化CO2/H2参与的C―H的甲基化反应99。在这项工作中,研究者证明了CO2和H2的组合可以用作甲基化试剂,用于(杂)芳烃的直接甲基化,如吲哚,吡咯和富电子芳烃,具有中等至良好的反应性(图27)。在反应的过程中,催化剂前体Ru(acac)2和配体triphos原位形成的催化活性物种是反应的关键,在此过程中酸共催化剂(譬如MSA)也是必需的。通过机理和控制实验发现原位生成的甲醛是构筑C―C键的关键步骤。在酸存在情况下形成阳离子[Ru-H]+催化活性物种,接着CO2被Ru―H还原形成Ru甲酸盐,随后将被碳亲核试剂进攻形成相应的缩醛中间体,最后缩醛氢解生成甲基化产物。

图27 Ru催化剂用于CO2、H2与杂芳烃的甲基化反应Fig. 27 Ruthenium-catalyzed methylation of heteroarenes with CO2 and H2.
木质素和CO2都是廉价易得的可再生的资源,使用木质素与CO2合成高值化学品是十分具有挑战的课题,并且对资源和环境的意义也很重大100。韩布兴等报道通过CO2、H2和木质素/芳基甲基醚在Ru-Co双金属催化体系作用下发生羰基化反应制备乙醇101。反应生成乙醇的TON能够达到145。一系列的控制实验揭示反应过程首先发生的RWGS,接着是C―C键的形成,最终得到目标产物乙醇(图28)。

图28 CO2、H2与芳基甲基醚(a)/木质素(b)制备乙醇Fig. 28 Synthesis of ethanol from aryl methyl ethers(a)/lignins (b) with CO2 and H2.
研究者又通过低温逆水煤气变换过程耦合CO羰基化的向高值化学品转化。反应的首先是在离子液HMimBF4存在的情况下,反应温度80 °C时,Ru3(CO)12就可以催化CO2逆水煤气变换生成CO,之后通过共催化剂催化CO羰基化反应。通过控制实验和同位素实验证实阴离子在RWGS的过程起到重要作用(图29)。研究者报道三种CO2羰基转化途径:1) CO2与二芳基醚反应生成具有药物活性氧杂蒽酮(Xanthone);2) CO2与苯甲醚反应生成重要的大宗化学品苯酚和醋酸;3) CO2与木质素反应生成醋酸102。
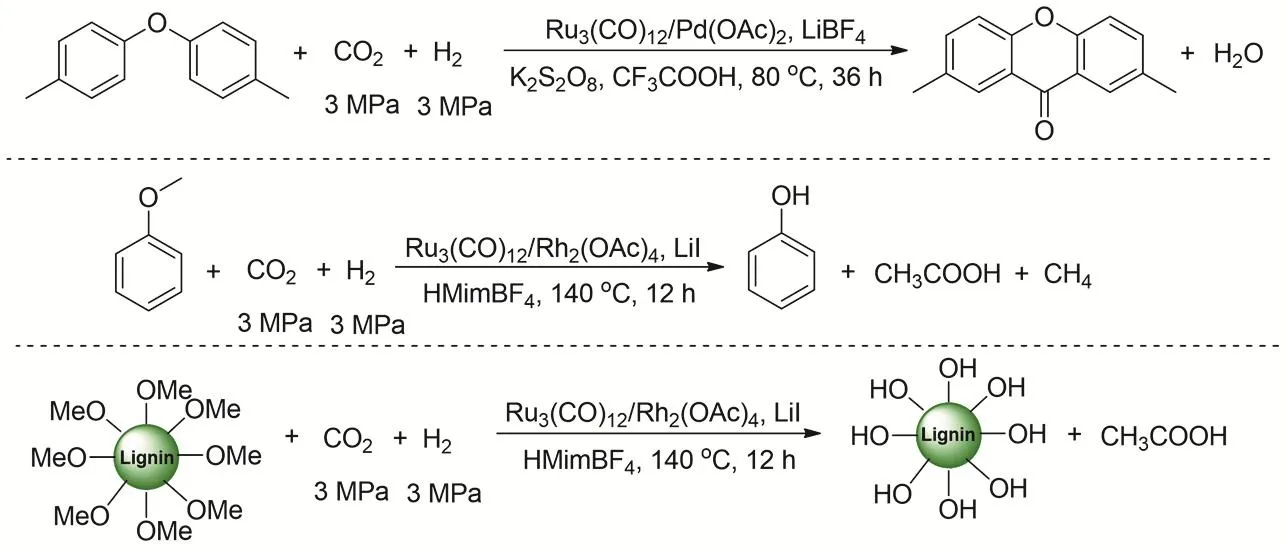
图29 可再生碳源向高值化学品转化Fig. 29 Transformation of renewable carbon resources to value-added chemicals.
3 总结与展望
CO2、H2参与的还原羰基化反应在热催化CO2还原领域的研究已经取得了一定进展。由于CO2和H2都是相对清洁、低成本的原料,可作环境友好的试剂生产更高附加值的化学品,而且缓解温室气体排放和碳资源的有效利用问题,因此是CO2化学转化利用研究最为活跃的前沿领域之一。然而,CO2上碳氧键的惰性,使金属活性物种加成到上面非常的困难,并且该反应过程常涉及多个竞争反应导致目标产物的化学选择性差,大量的研究仍处于实验室研发阶段。因此,积极开发新型稳定高效催化体系和经济可行的工艺路线,对实现CO2还原羰基化技术的工业化应用依然是本领域的重要研究方向。我们认为将来的研究主要可以从以下两个方面开展。
1) 发展更加高效的催化体系、非贵金属体系甚至无金属催化体系。均相催化CO2还原羰基化的过程,首先是CO2分子活化的过程,常用贵金属催化体系效率偏低的根源是不能对惰性的CO2分子有效的活化。通过在催化剂上设置能够活化C=O的配体官能团,结合金属催化中心以协同催化的方式完成对CO2分子的还原,提高金属催化剂的原子利用率,减少催化剂用量。针对该类反应是多个反应的串级过程,可以发展两元/多元金属催化体系,利用它们各司其职或协同催化完成整个过程。另一方面,通过催化体系合适配体/添加剂促进金属催化剂对CO2分子的精准活化。此外,非贵金属络合物,如Fe、Co、Mn等,催化的CO2化学转化也是该领域值得关注的发展方向。
2) 利用现代表征手段和理论计算探究反应的机制。均相催化CO2还原羰基化反应是一个反应路径较为复杂的化学过程,涉及多个串级的反应以及一系列的竞争反应。每个反应对应的不同的催化活性物种,也是造成催化剂用量高的主要原因。因此,有效结合现代表征手段(如原位红外光谱、电喷雾电离质谱和原位同步辐射等),追踪和捕捉反应过程活性物种的形成和中间体,探索催化剂原子利用率不高的内在原因,有助于对均相催化CO2还原羰基化反应机理的深入认识。同时,利用理论计算对反应的中间路径进行模拟,有针对性的找出反应的瓶颈问题所在,为催化体系的设计提供可靠的参考。总之,随着研究的不断深入,不但可以高效催化CO2转化制备高值化学品,而且实现碳的化学循环,成为自然界碳循环的有力补充。
猜你喜欢
化工管理(2022年13期)2022-12-02
中国饲料(2021年17期)2021-11-02
石油炼制与化工(2021年3期)2021-03-23
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
航天工业管理(2020年9期)2020-12-28
化工时刊(2020年7期)2020-09-04
化工技术与开发(2020年8期)2020-08-26
重型机械(2020年2期)2020-07-24
化工时刊(2020年11期)2020-01-12
中国特种设备安全(2019年9期)2019-12-03
