
光催化CO2还原的超薄层状催化剂
2021-06-02 11:38秦祖赠吴靖李斌苏通明纪红兵
物理化学学报 2021年5期
秦祖赠,吴靖,李斌,苏通明,纪红兵,2,*
1广西大学化学化工学院,南宁 530004
2中山大学化学学院,广州 510275
1 前言
二氧化碳是化石燃料燃烧的最终产物。工业革命前,大气中CO2浓度在300 μL·L-1左右。200年来,随着科技的发展和人口的暴增,化石燃料的消耗越来越大,CO2排放量持续增加,目前大气中CO2的浓度达到400 μL·L-1以上1。二氧化碳的浓度增加对人类的生存环境和地球的生态系统造成严重影响,因此,探索如何减少大气中CO2的含量并合理利用CO2已经成为全世界重要研究课题之一。解决这个问题的理想方案之一是通过光催化将二氧化碳转化为可利用化学品2。太阳光作为唯一的能量输入,虽然效率很低,但已成功在光催化系统中将CO2转化为CH43、CO4、CH3OH5、HCOOH6及其它可利用化学品。在过去的几十年中已经探索了大量用于CO2还原的半导体或复合半导体光催化剂,包括TiO27、CuO8、ZnO9、WO310、BiYO311,12、Bi2WO613、NaNbO314、BiVO415等金属氧化物,ZnS16、CdS17、CdSe18、CoTe19等金属硫属元素化物,GaN20、Ni2P21等金属氮化物和磷化物,ZnAl-LDHs22等层状双氢氧化物和非金属半导体,如类石墨相氮化碳(g-C3N4)23、SiC24、氧化石墨烯(GO)25和碳量子点(CQD)26等。
但大多数半导体的光催化效率相对较低,不能满足大规模工业应用,主要原因如下:(1) 光吸收范围较小:传统的半导体带隙偏大,只能被具有较高能量的紫外光激发,而紫外光仅在太阳光谱中占4%-5%;(2) 光生载流子的分离和迁移效率低:在迁移到半导体表面的过程中,大部分光生电子和空穴会发生复合,大大降低了光催化性能;(3)反应活性位点不足:由于比表面积不足,仅有少量光生载流子能到达催化剂表面与少量吸附在表面CO2进行反应。
然而,超薄层状材料的出现为提高CO2还原光催化活性带来了前所未有的机遇。超薄层状光催化材料不仅可以改进块状光催化材料固有的物化特性,而且还可以产生块状催化材料没有的新特性,如:超高比表面积和大量低配位表面原子将有利于反应物CO2的吸附和活化27;低配位表面原子更容易从晶格脱离形成空位缺陷,有利于CO2吸附和电子结构的改善;超薄厚度可导致明显的量子限域效应,而引起导带(CB)和价带(VB)位置变化及带隙变宽,可提高氧化还原能力28,29;超薄的平面结构大大缩短了光生载流子从内部迁移到表面反应部位或界面的距离,从而减少了光生载流子的损失;与跨层的载流子迁移相比,载流子在同一层中沿着表面移动要容易得多,可确保沿表面的高电荷迁移率30;2D结构有利于催化剂与其他组成单元更紧密地组装或生长,是复合光催化剂的理想基材;由于2D结构内部原子占比较小,只需要通过表面掺杂、修饰等手段就可以大大改变催化剂的特性和性能。
基于上述优点,具有单层或少层厚度和清晰结构特征的超薄层状材料可以在结构和性能之间建立精确的关系,这为获得高效、稳定的CO2还原光催化剂提供了理论依据,而且在促进光催化CO2还原机理的研究中起着重要的作用31。本文系统地总结用于光催化CO2还原的超薄层状催化剂的最新进展:首先简单介绍光催化CO2还原的基本原理;其次对用于CO2光催化还原的超薄层状催化剂进行分类;然后介绍层状催化剂的制备方法,重点对超薄层状光催化剂光催化CO2还原性能的改良策略进行总结;最后,对超薄层状光催化剂在CO2还原领域的未来前景和挑战进行了展望。
2 光催化还原CO2的基本原理
半导体光催化还原CO2的过程如下:首先在光子能量大于或等于带隙的光线照射下,催化剂价带(VB)上的电子被激发跃迁到导带(CB)上,产生光生电子-空穴对。随后光生电子-空穴分离并转移到光催化剂表面,这一过程中光生电子-空穴可能发生复合而消耗掉。最后迁移到表面的光生电子与CO2和水中的H+发生还原反应生成有机化合物(导带电位要比表面电子受体电位更负),而光生空穴则与水发生氧化反应生成O2(价带电位要比表面电子供体电位更正)从而完成整个光催化反应。
目前研究进行的光催化活性测试主要分为气-固反应和气-液-固反应,不同的反应条件得到的产物也会有区别。例如,层状钙钛矿Bi2MoO6在气-固反应条件下的主产物是CO,而在气-液-固三相反应中的主产物则是甲醇。另外,除了甲酸和乙醇由气液固反应产生之外,其他种类产物与反应相态并无直接的联系,而与催化剂的种类,导带位置密切相关。更负的导带位置意味着更强的还原能力,可以克服更高的动力学障碍,但这也意味着带隙的增大,电子跃迁所需能量更大。因此,调整光催化剂能带结构会大大影响产物的产率和选择性。通过还原CO2来产生CO和HCOOH是一种双电子过程,该反应具有相对较低的动力学障碍。CH3OH和CH4的形成是6、8电子过程,包含多步中间反应以克服更高的动力学障碍,这些过程的机制仍在研究中。上述反应式和反应电极电位如式(1)-(7)所示。

3 光催化CO2还原层状光催化剂的分类
3.1 非金属层状光催化剂
3.1.1 石墨相氮化碳
石墨相氮化碳(g-C3N4)是最具代表性的非金属层状光催化剂,主要由碳和氮构成的共价固体,其中三嗪环和三-s-三嗪环作为其基本组成单元(图1)32。通过聚合各种富氮材料如三聚氰胺33,尿素和硫脲34等,可以方便地合成该物质。g-C3N4是一种非常稳定的有机材料。它可以在高温下保持稳定性(最高可达到600 °C)。但也发现将所得的g-C3N4在空气中退火至550 °C会导致g-C3N4剥落,BET表面积增加,带隙增大35。除了低成本外,g-C3N4在CO2光催化还原方面的优势还在于:(1) 富含有利于CO2活化的氮原子;(2) 有利于光催化还原CO2为各种增值化合物的能带结构;(3) 二维层状结构有利于电子转移到催化剂表面吸附位点;(4) 通过改变前体和聚合条件,可以获得具有略微不同带位置的g-C3N4;也可以通过用其他非金属掺杂来大大改变g-C3N4的光吸收和电子转移能力。简单地将大量g-C3N4剥离成几层纳米片将促进电子转移到表面活性位点并改变光催化CO2还原中的产物选择性。将带隙为2.77 eV的块状g-C3N4热剥离为带隙为2.97 eV的纳米片,主要产物会从CH3CHO变成CH435。猜测g-C3N4纳米片电子转移和扩大的表面积有助于产生CH4和CH3CHO的氧化。目前纯g-C3N4光催化剂的主要问题是可见光利用率低,电导率差,电子和空穴复合率较高。它在420-460 nm的光催化反应量子效率仅为0.1%36,这远远不能满足实际应用的需要,因此需要通过各种手段来改良其催化性能。
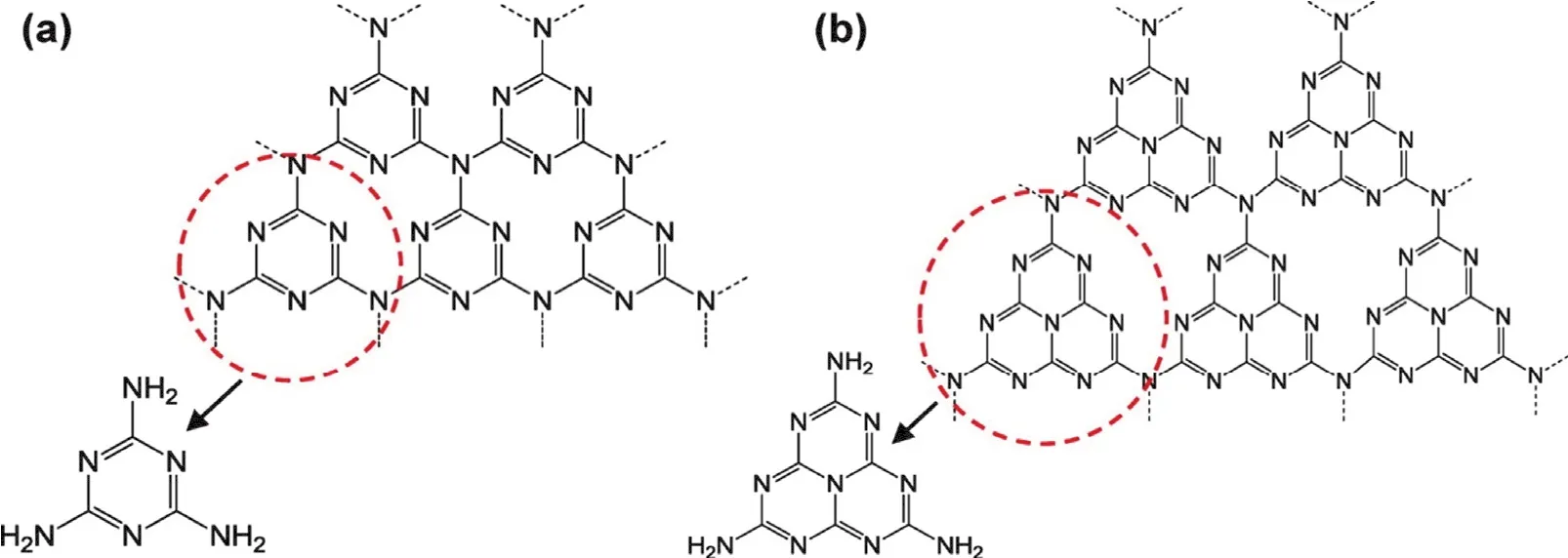
图1 (a) g-C3N4的三嗪和(b)三-s-三嗪结构32Fig. 1 (a) Triazine and (b) tri-s-triazine structures of g-C3N4 32.
3.1.2 黑磷
黑磷(BP)是除白磷和红磷以外的磷的单质第三种同素异形体,也是最稳定的形式之一。BP具有类似于石墨烯的2D分层结构,块状BP由许多单层BP通过弱的范德华力堆叠,并且每个连续层由具有sp3杂交的P4单元形成。这种杂交导致非平面折叠的六边形结构与褶皱的蜂窝状结构相似。这种特殊的结构会破坏P4的各个键,从而生成sp3杂化,两个不同的键角分别为96.300° (01)和102.095°(02),从而使BP更加稳定(图2)37。2014年,使用与石墨烯相同的“粘胶带剥离技术”制备了单层黑磷(BP)38,它表现出优秀的光电性能和光吸收能力39,40。这些性质都是光催化过程非常需要的。
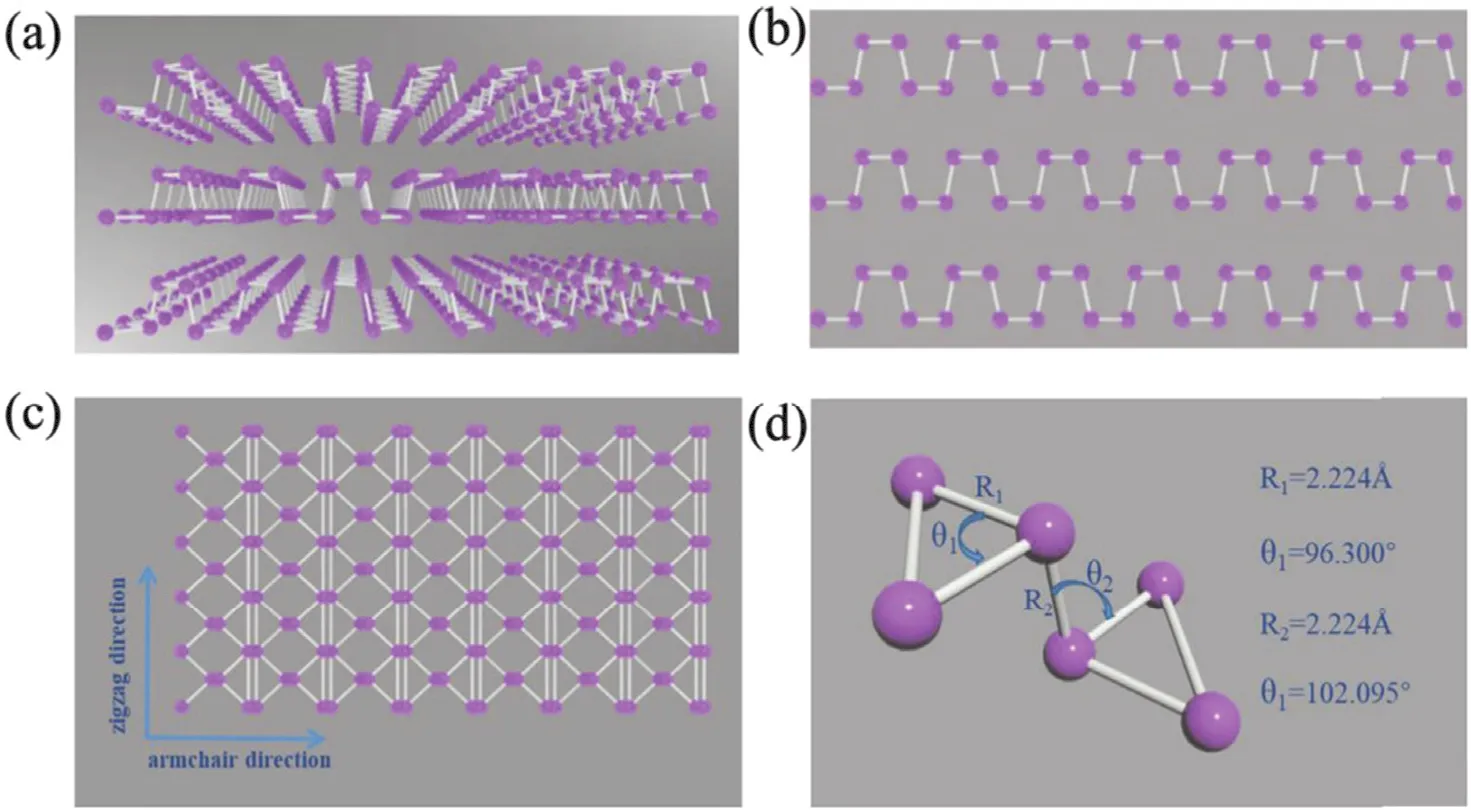
图2 (a) 层状BP的晶体结构,(b) 层状BP的正视图,(c) 层状BP的垂直视图,(d) 层状BP的详细晶格参数37Fig. 2 (a) The crystal structure of layered BP, (b) the front view of layered BP, (c) the vertical view of layered BP, and (d) the detailed lattice parameter of layered BP 37.
黑磷的稳定性一直是困扰研究人员的一大难题,它在空气中非常容易氧化,导致性能降低,通过表面修饰的方法可以大大缓解这一现象41。由于阳离子-π相互作用,在溶剂中自由分散的银离子可以自发的吸附到黑磷的表面,钝化黑磷中磷原子的孤对电子,进而极大提高了黑磷片层的稳定性。
块状BP的实验带隙为0.35 eV42,43,而单层BP的带隙为1.6 eV44,带隙的增加高度依赖于层数减小(图3)44。紧束缚理论,赝势和狭义的第一性原理计算已揭示了大块BP和单层BP的能带结构在可见光或红外区域能带结构类似于p型直接半导体。由于其较低的CB位置(较高的还原能力) (约-0.9 eV),BP也是一种用于光催化还原CO2的有前途的光催化剂。最近,叶立群教授的小组首先通过简便的静电吸引方法将BPQD固定在g-C3N4上进行光催化CO2还原实验45。BPQDs含量为1%时,最佳 CO 产 率 为 6540 μmol·g-1·h-1, 同 时 有 290 μmol·g-1·h-1CH4生成,这比纯g-C3N4大得多。上述结果表明,BPQD是增强BPQDs/g-C3N4催化活性的优良光催化剂。催化循环测试表明,在相同条件下,连续几次循环实验后,CO生成的损失可以忽略不计,XPS结果揭示了BPQDs/g-C3N4纳米复合材料在可见光照射下相对稳定的原因,因为形成的氧化层和P-N配位键阻止了BPQDs在可见光、氧气和水条件下的分解。
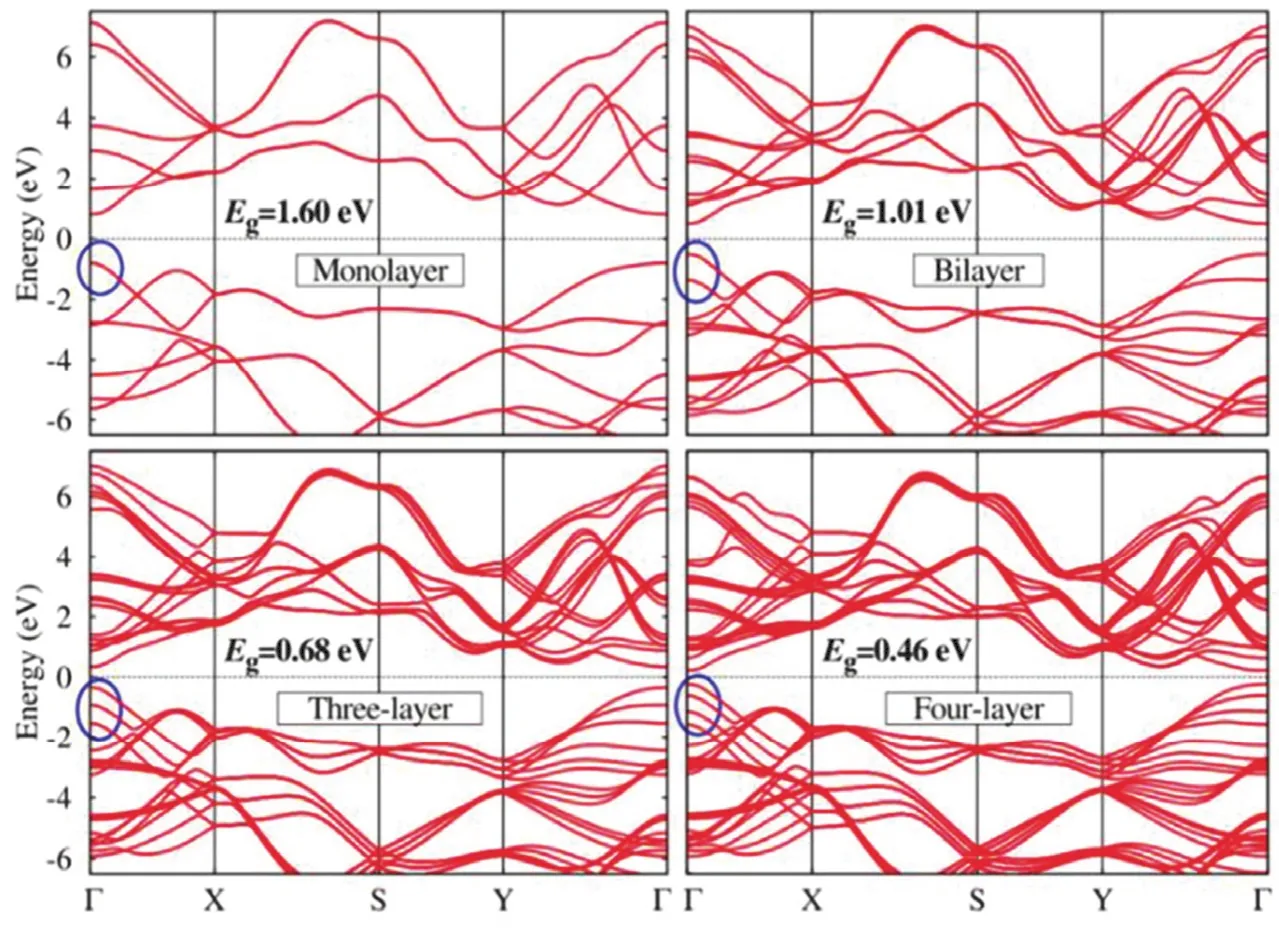
图3 在GW方法中n = 1-4计算的n层BP的能带结构44Fig. 3 Band structures for n-layer BP calculated within the GW approach for n = 1-4 44.
目前实验室制备少层或单层黑磷主要的方法是液相超声法,这种方法虽然可以少量剥离黑磷,但黑磷分散液的浓度,剥离的大小,层数始终无法很精确的控制,且效率很低。因此,黑磷纳米片的高效可控制备是目前黑磷开发的一个难点。
3.1.3 氧化石墨烯
氧化石墨烯(GO)作为从氧化石墨上剥离下来的单层材料,仍保持石墨的层状结构,但在每一层的石墨烯单片上引入了许多氧基功能团(图4)46。GO的基面被环氧化物和羟基共价包围,而边缘被羧基官能团修饰47。含氧官能团的引入将一部分sp2碳转化为sp3。氧含量的增加促进了导电sp2和非导电sp3域共存,从而导致带隙的增加48。GO由于其较低的电子迁移率而表现为p型半导体。通过可控的氧化过程,GO带隙在2.4-4.3 eV之间可调49,这为光催化应用提供了可能性。例如,使用改良的Hummer方法制备的改性氧化石墨烯(GO-3)在可见光下光催化CO2转化为甲醇的速率为0.172 μmol·g-1·h-1,是纯TiO2的六倍50。出色的光催化性能归功于催化剂制备过程中H3PO4的加入的量足够,保护GO基面免受过分氧化,并显著提高了CO键的强度。另一方面,C=O和-COOH官能团含量则较低,从而缩小了带隙。
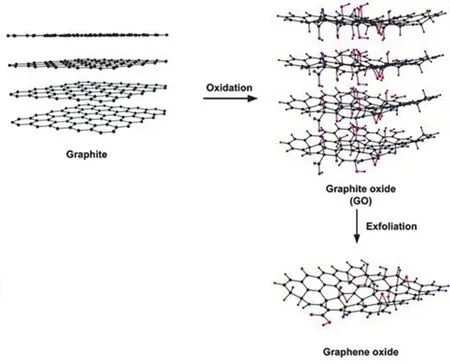
图4 用Hummers法合成氧化石墨烯46Fig. 4 Synthesis of graphene oxide by Hummers method 46.
3.2 含金属层状光催化剂
3.2.1 金属氧化物纳米片
迄今为止,金属氧化物是研究最广泛的光催化剂。由于2D材料具有更大的比表面积,激子迁移效率,近来金属氧化物纳米片的研究也多了起来。已经控制了几种具有超薄2D结构的金属氧化物的制备,并将其应用于光催化CO2还原应用,例如TiO251,ZnO52,WO353和Co3O454等。与块状材料不同,超薄纳米片具有几乎100%的表面结构,暴露在表面上的原子与材料内部的原子表现出不同的化学状态,因此它们表现出不同寻常的特性。利用Co-MOF-NS作为前驱体,成功合成了超薄Co3O4纳米片用于高效光催化还原CO2(图5)54。得益于MOFs前体的结构性质,煅烧后的Co3O4-NS继承了2D的形态和发达的孔隙度,这增加了催化剂的比表面积,提供了更多的CO2吸附和活化位点,缩短了载流子转移到催化剂表面的距离,利于载流子的分离和输运。DFT计算表明,单层Co3O4(-0.27 eV)具有比块状Co3O4(-0.13 eV)更强的CO2吸附能,这有利于CO2转化成CO。结果,CO产生速率为约为4.52 μmol·h-1,是块状Co3O4催化剂(Co3O4-BK)的1.8倍。

图5 超薄多孔Co3O4纳米片的合成过程示意图Fig. 5 Diagram of the synthesis process of ultra-thin porous Co3O4 nano sheet.
3.2.2 层状钙钛矿
层状钙钛矿半导体由不连续的钙钛矿板交替堆叠而成,具有无限延伸的钙钛矿结构和高稳定性,已被大量研究用于光催化CO2还原(图6)55。沿层状钙钛矿的c轴方向,间隔层产生绝缘性能,电子迁移率低,产生了各向异性的电子转移能力,这为层状钙钛矿带来了特殊的光电性能55。典型的Aurivillius相钙钛矿如Bi2WO656和Bi2MoO657,具有2.6-2.7 eV的窄带隙,并倾向于形成由大量纳米板构成的复杂类花结构,这种结构非常有利于可见光驱动的二氧化碳还原。具有良好的CO2吸附和光还原性能的Sr3Ti2O758属于Ruddlesden-Popper层状钙钛矿。对于这种结构的钙钛矿,(SrTiO3)2钙钛矿层夹在(SrO)间隔层之间,位于钙钛矿界面的Sr阳离子与一侧八个相邻的氧阴离子和另一侧一个氧原子呈不对称配位。另外,在紫外光下光催化CO2还原活性出色的ALa4Ti4O15(A = Ca, Sr, Ba)59属于(111)层状钙钛矿,这种结构的钙钛矿属于A位金属阳离子和角共享BO6八面体形成的(111)钙钛矿板构成的一系列同源层状结构。
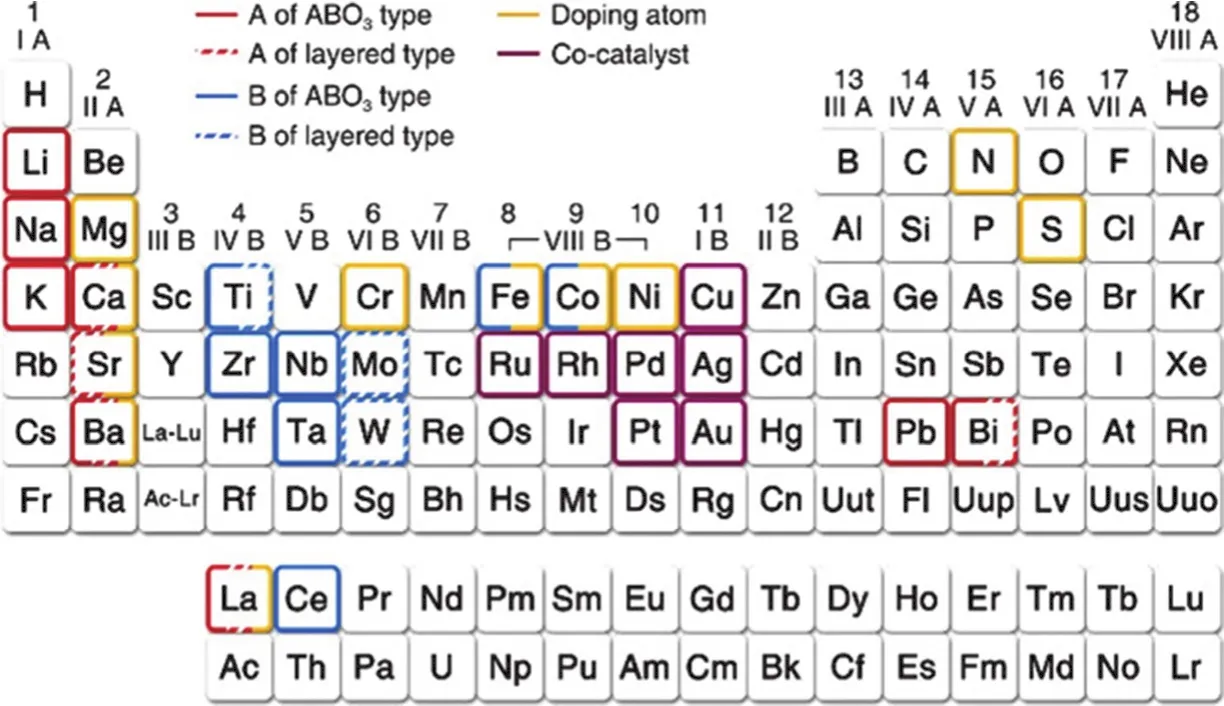
图6 用于光催化CO2还原的钙钛矿光催化剂的元素组成55Fig. 6 The element composition of perovskite photocatalyst for the reduction of CO2 55
3.2.3 卤氧化铋
近年来,具有特殊层状结构的V-VI-VIII三元氧化物半导体卤氧化铋(BiOX,其中X = Cl, Br, I)由于其出色的光电性能,化学稳定性,环保和廉价而在光催化领域得到了广泛的研究60,61。BiOX的层状结构可提供足够的空间来极化相关的原子和轨道,从而激发[Bi2O2]与卤素平板之间的内部电场(IEF)的形成。并且由于诱导的IEF可以加速光激发电子-空穴对的分离和迁移,BiOX的光催化活性得到了显着改善62。此外,BiOX的VB顶部和CB底部分别位于R和Z点,表明它们是间接带隙半导体。也就是说,BiOX上的光致电子应从VB经过一定的k空间距离才能到达CB,从而进一步降低光致电子与空穴的复合率63。BiOCl纳米片仅在紫外光下有CO2光还原活性64,而BiOBr纳米片和BiOI纳米片在可见光下也能产生作用65,66。为进一步增强光催化能力通过乙二醇辅助溶剂热法制备了含氧空位的BiOBr纳米片以改善光催化还原活性67。所制备的含氧空位的BiOBr纳米片在可见光照射下获得了4.86 μmol·g-1的CH4总产率,模拟太阳光照射下CH4的总产率为9.58 μmol·g-1,而原始BiOBr这两个产率仅为1.58和2.99 μmol·g-1。表面氧空位缺陷的存在导致BiOBr带隙变窄,增强了光吸收以促进电子-空穴对的产生。同时,氧空位对捕获光诱导电子起着不可或缺的作用,从而抑制了光生载流子的直接复合。除此之外,CO2分子与氧空位之间的吸引显著降低了界面电荷转移的能垒。值得注意的是,在CO2光还原过程中氧空位会再生,因此含氧空位的BiOBr具有长期稳定的催化活性。
3.2.4 过渡金属硫化物(TMDC)
由于独特的电子结构,过渡金属硫化物通常可显示出相对较宽的光吸收区域,这被认为是光催化应用中的一类有前途的材料。迄今为止,大约有40种2D TMDC类型,通常缩写为MX2,其中M主要是从IVB组到VIII组的过渡金属元素,X包括VIA组的S,Se和Te的非金属元素(图7)68。TMDCs纳米片呈三明治形式,MX2分子层是通过将M原子层夹在两个六角形X原子层之间形成的,可以用“X―M―X”表示。目前已经制备出许多超薄二维金属硫属化合物,并显示出出色的光催化CO2还原性能,包括MoS269,SnS270等。以MoS2为例,尹晓红教授等用水热法合成了花状的MoS2纳米片,随后在锂离子存在下在邻二氯苯溶液中超声制造出了层状MoS269。为了提高光催化性能,将银纳米粒子与准备好的MoS2纳米片复合。当Ag在MoS2上占20% (w)时,甲醇的最高收率达到365.08 μmol·g-1·h-1。和CdS一样,这类材料存在着一个通病:易被光生空穴氧化71。因此,需要采取复合,加牺牲剂等各种手段来阻止这一过程。
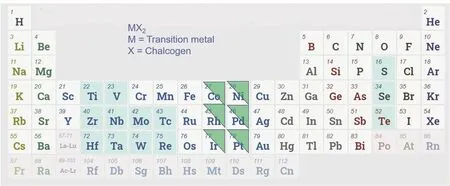
图7 约40种层状TMDC含有的过渡金属和硫族元素在周期表中的彩色显示Fig. 7 Transition metals and sulfur group elements contained in about 40 kinds of TMDC displayed in color in the periodic table.
3.2.5 MXene材料
MXene是一种具有类似于石墨烯结构的层状过渡金属碳化物或碳氮化物,化学通式为Mn+1XnTx(n = 1, 2, 3),其中M是过渡金属元素,X是碳或氮元素,Tx表示通过化学蚀刻前体MAX相而产生的官能团(-OH,-F或-O)72。目前成熟的制备MXene技术是通过HF对A元素进行选择性蚀刻。大多数MXene由于其金属特性而无法生成光载流子,只能作为助催化剂用于改善其他催化剂的催化性能72,73。只有一些具有窄带隙(约1.28至2.96 eV)且负CB电位大于CO2还原电位的MXene材料如Ti2CO2、Hf2CO2和Sc2CO2可能具有CO2光还原活性74。DFT计算表明,Ti2CO2可以对光催化CO2还原生成HCOOH有很高的选择性(图8)。另外,由于CO2在完美的端氧表面上的吸附太弱,发现Ti2CO2上的氧空位(Ov)对提高CO2的吸附有重要作用75。
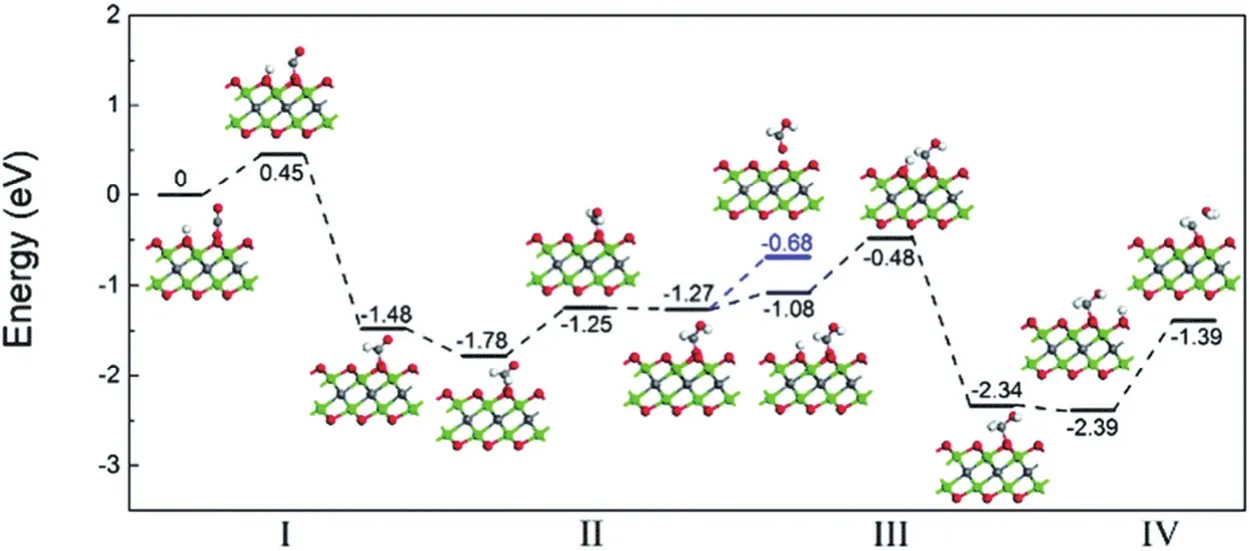
图8 Ti2CO2单层中Ov处CO2加氢的反应途径Fig. 8 Reaction pathway of hydrogenation of CO2 at Ov in Ti2CO2 monolayer.
3.2.6 层状双氢氧化物(LDHs)
通过掺入特定的光敏金属阳离子,可以在金属氢氧化物中可控地调节带隙76。因此,具有超薄2D结构的层状双氢氧化物在光催化应用方面显示出巨大的潜力。例如,典型的层状双氢氧化物ZnAl-LDH超薄纳米片可用于将CO2光还原成CO (图9)22。TEM图像显示纳米片厚度约为2.7 nm,对应于2个LDH层的厚度。得益于超薄厚度,ZnAl-LDH超薄纳米片中产生了氧空位(Vo),Zn+-Vo络合物可作为捕集位点以有效吸附CO2和H2O分子,从而促进光诱导的电荷分离并显着提高CO2光还原的活性。DFT计算表明,ZnAl-LDH超薄纳米片相对于块状ZnAl-LDH的带隙中出现了一个新的缺陷能级,该能级同时受到Zn 4s轨道和O 2p轨道的贡献。这进一步证实了Zn 4s轨道电子的存在,与ESR和EXAFS数据表明的Zn+-Vo配合物一致。
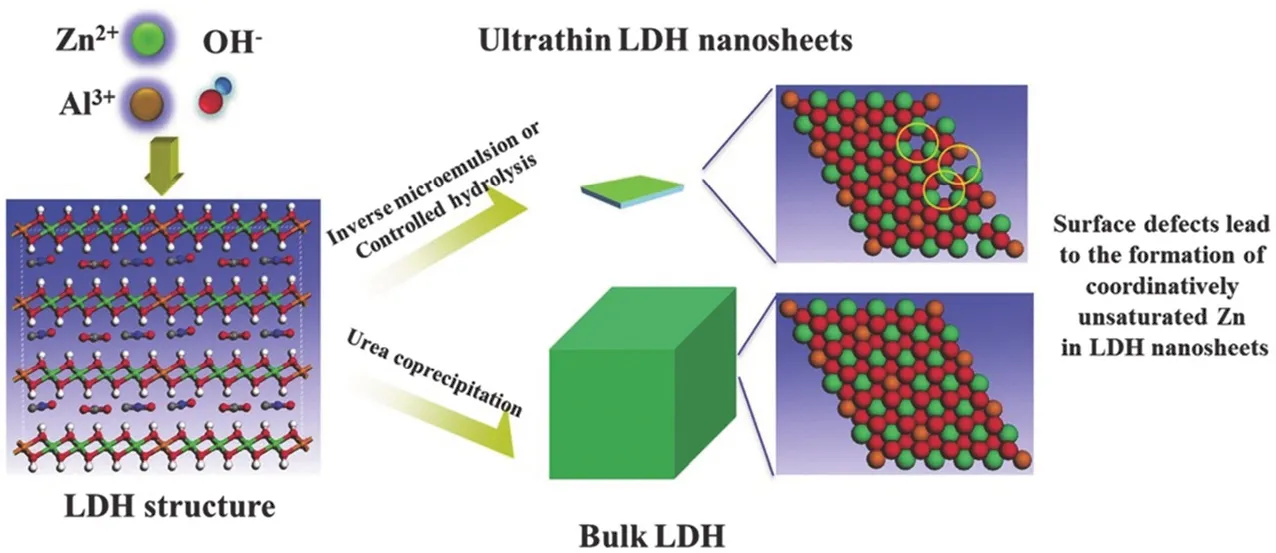
图9 配位不饱和的ZnAl-LDH纳米片的形成示意图22Fig. 9 Formation of coordination unsaturated ZnAl-LDH Nanosheets 22.
3.2.7 二维金属有机骨架(MOF)
MOFs是一个有趣的结晶多孔固体家族,由无机金属离子(或簇)和有机桥接配体构成,具有大的表面积,高孔隙率和良好的CO2分子捕获能力,以及灵活可调的结构组成和功能性质77。MOFs材料已广泛在气体存储,分离和非均相催化领域应用。MOFs结构中的金属-氧簇可视为通过有机配体连接的离散半导体量子点,通过配体-金属电荷跃迁,配体上的光生电荷可以转移到催化位点78。该电荷跃迁类似于半导体中的带隙,其宽度随有机配体和金属中心的种类而变化。Ye等开发了一种以二维Zn-MOF纳米片为半导体光敏剂,以[Co2(OH)L](ClO4)3或ZIF-67为助催化剂的体系,用于CO2的光还原79。这种MOF/ZIF体系是首次开发出来的,而Zn-MOF纳米片也是首次应用到CO2光还原中。与块状Zn-MOF相比,采用Zn-MOF纳米片作为光敏剂时具有更高的光催化效率和选择性。光电化学阻抗和光致发光研究表明,Zn-MOF纳米片的光催化增强主要是由于其具有更好的电荷输运能力、更有效的分离和更长的寿命。这项工作为开发各种2D-MOF基光催化材料提供了机会。
Wang等通过DFT计算系统地研究了超薄的二维基于钴卟啉的Co-O簇MOF(Co-PMOF)对CO2还原的光催化性能80。DFT计算表明,Co-PMOF单层催化的从CO2到CH4的最佳还原路径是:CO2→*COOH → *CO → *CHO → *CH2O → *CH2OH →CH3OH → *CH3→ CH4。在Co-PMOF单层上,速率控制步骤从*CHO到*CH2O的ΔG仅为0.39 eV,CB最低值比CO2/CH4电位高-0.53 V,说明CoO簇与钴卟啉的结合明显促进了CO2的还原过程。在不引入任何助催化剂的情况下,2D Co-PMOF可以作为一种很有前途的光催化剂直接将CO2还原为CH4。
MOF材料的水稳定性一直是困扰研究人员的一大难题。另外,这类材料大多只有还原CO2的能力而没有氧化H2O的能力,需要借助牺牲剂来消耗空穴,目前用MOF进行的光催化CO2还原反应几乎都在乙腈的三乙醇胺溶液中进行81。因此,开发水稳定且高CO2还原活性的MOF还需要付出非常大的努力。
3.2.8 二维层状锗硅烷
硅烯和锗烯是与石墨烯类似的第四类二维Xenes,直接带隙分别为1.55和23.9 meV82。它们比石墨烯具有更好的带隙可调性。因此它们在催化领域具有巨大的潜力83。最近,封伟教授等在这方面的研究上取得重要突破84:在理论计算和结构设计的基础上,采用-H/-OH封端二元锗硅烯,首次获得了带隙可调控的二维层状锗硅烷HGeSiOH(图10c)。通过将硅含量由10%提高到90%,其带隙可从1.8 eV增加到2.57 eV (图10a,b)。此外,合适的能带位置赋予了这种材料将CO2光还原为CO的能力。当硅锗比为1 : 1时,合成的HGeSiOH具有较宽的光谱响应范围和合适的能带位置,最有利于电子和空穴的光生。样品相对较高的比表面积(319.7 m2·g-1)有利于光生载流子的迁移。DFT计算表明,HGeSiOH的结构增强了材料对CO2的吸附能力,VB和CB杂化轨道组成和分布有利于光生载流子的迁移和抑制光生电子与空穴的复合。此外,表面氧空位缺陷的存在导致了电子的富集,增强了CO2分子的活化,有利于CO2的还原。结果,HGeSiOH在300 W氙灯下CO的产率为6.91 mmol·g-1·h-1,AQE高达5.95%,优于大多数最近报道的光催化剂。
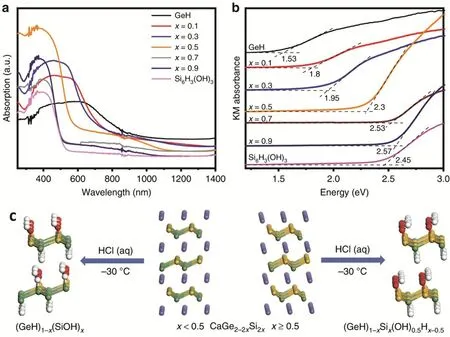
图10 锗硅烷(x = 0.1, 0.3, 0.5, 0.7, 0.9),GeH和Si6H3(OH)3的(a)紫外-可见漫反射光谱和(b) Tauc图;(c) CaGe2-2xSi2x到锗硅烷(GeH)1-x(SiOH)x (x < 0.5)或(GeH)1-xSix(OH)0.5Hx-0.5 (x ≥ 0.5)的拓扑分离示意图 84 Fig. 10 (a) UV-Vis diffuse reflectance spectra and (b) Tauc plots of gersiloxenes (x = 0.1, 0.3, 0.5, 0.7, 0.9),GeH and Si6H3(OH)3; (c) schematic illustration of topotactic deintercalation of CaGe2-2xSi2x to gersiloxenes(GeH)1-x(SiOH)x (x < 0.5) or (GeH)1-xSix(OH)0.5Hx-0.5(x ≥ 0.5) 84.
4 超薄层状材料的制备
4.1 剥离或刻蚀块状材料
由于过去的研究中大多数制备方法获得的层状材料都是通过范德华相互作用而堆叠在一起的块状,因此剥离或刻蚀的方法已广泛用于制备超薄的层状材料。堆叠的层状材料由于外力作用失去相邻层之间的相互作用,层与层分裂而获得超薄纳米片。在此,我们将介绍常用的剥离和蚀刻方法,包括机械剥离、液体剥离、气体剥离和化学蚀刻。
机械剥离法是制备超薄层状材料最早、最常用的合成方法。2004年,英国曼切斯特大学物理学家Novoselov和Geim采用机械剥离法首次成功将单个石墨原子层从块体石墨当中分离出来85。这标志着单原子层材料的诞生,表明单个原子层的材料是可以稳定存在的。而由于这项工作对凝聚态理论的颠覆以及对2D材料研究产生的重大意义,这两位发现者在2010年共同赢得了诺贝尔物理学奖。2014年,Liu等人第一次通过粘带微裂解法制备了单层或多层黑磷38。通过该方法获得了高质量的2D黑磷,但收率很低。另外,粘合剂残留物也会导致黑磷被污染。Castellanos-Gomez等通过使用胶带与粘弹性聚二甲基硅氧烷偶联来剥离母体BP的方法,对这种常规方法进行了改进,这项技术极大地提高了通量并消除了BP的污染86。然而,机械剥落需要多步操作(粘合和剥离),并且制成的薄片的尺寸和厚度不可控,这需要其他设备来确定,例如原子力显微镜87。此外,该方法的处理量相当低,不能满足研究和实际应用的要求。并且此路线仅适用于大型和分层的散装材料。
液体剥离是制备原子薄的层状材料的简便,可扩展,可控且对环境友好的方法之一88。主要是通过引入液体中的分子/离子到层间增加层间距,减少层间相互作用,然后在超声波处理下导致层间剥离,最终形成稳定的胶体溶液。通过这种方法已经成功地制造了许多2D材料,例如石墨烯89,90,g-C3N491,92和氮化硼(h-BN)93。2014年,Brent等人首先提出了一种简单且可扩展的方法,以N-甲基-2-吡咯烷酮(NMP)为溶剂,制备尺寸为200 nm ×200 nm的三至五层黑磷。随着剥落时间的延长(长达48 h),可以获得尺寸为20 nm × 20 nm的单层和双层黑磷94。一年后,有研究发现具有惰性和极性的二甲基甲酰胺(DMF)和二甲基亚砜(DMSO)是制造原子级薄黑磷的合适有机溶剂95。所制备的产品高度分散且具有良好的抗降解性,并且机械剥离的BP表现出优异的电性能。
气体剥离法是近些年新兴的层状材料剥离技术,具有无污染、产率高的优点。2016年,一种用N2制备少量层状h-BN的气体剥离法被开发出来96。首先通过800 °C高温处理使大块h-BN的层间距因材料膨胀而增大,从而削弱层间范德华力相互作用。随后加热后的h-BN被抛入低温液体N2(-196 °C)中后,温度的巨大变化将导致大块h-BN的卷曲和分层。同时,剧烈膨胀的低温液态N2转变为气态N2,沿裂隙从卷边进入夹层。经过10次循环后,可以获得少于5个原子层的纳米片,质量产率高(16%-20%)。这一方法的关键在于将高温引发的原材料膨胀和低温液体气化相结合,对原材料进行去角质处理。通过调节预煅烧温度、低温液体种类和循环次数,可以获得不同的剥离效果。这一有效的制备方法有望推广到其它由范德华力连接的层状催化剂的制备中。
通过化学蚀刻的方法,也可以获得超薄材料。MXene是最典型的采用蚀刻制备的一类层状材料。为了详细说明MXene,我们必须了解一个特殊的层状MAX相族。首字母缩写MAX来自于材料的成分Mn+1AXn(n = 1, 2, 3),其中M代表早期过渡金属,如Sc、Ti、Cr、Nb、V、Mo等,‘A’是基团IIIA或IVA元素,‘X’代表C或N。MAX相对较强的金属M-A键表明,机械方法不适合剥离Mn+1Xn层。然而,M-A和M-X键之间的明显化学反应性使得选择性刻蚀成为可能。因此绝大多数MXene都是用HF刻蚀的方法合成的97。除了MXene之外,石墨氮化碳也可以用化学蚀刻的方法减小厚度。通过高温反复热氧化刻蚀,g-C3N4纳米片的厚度可以进一步降低到0.64 nm98。
4.2 直接合成超薄结构
尽管可以通过剥离和刻蚀方法来制备超薄的层状材料,但这类方法仅适用于层状材料。同时,这类方法也存在一些不足,如产量低,层数难以控制,成本高。因此直接合成的方法似乎是超薄层状材料制备的首选。这类方法具有更宽的材料适用范围,更高的产率,产品质量均一,厚度可控。这里介绍几种常用的合成超薄结构方法,包括化学气相沉积(CVD)、模板法、表面活性剂自组装法和层状杂化中间体方法。
化学气相沉积(CVD)法作为制备高质量层状材料的有效方法已经被广泛应用99,100。可以通过MOCVD技术在250 °C至450 °C的温度下将二氧化钛薄膜沉积在钠钙玻璃衬底上101。XRD显示所有膜具有纯的锐钛矿相。随着沉积温度的升高,平均粒径减小并且质地改变,光催化性能减弱。Meier等采用CVD法合成了由大量MoS2纳米片组合成的纳米花并将其应用于光催化CO2还原反应中102。光催化产物CO随着纳米花“花瓣”密度的增加而增加,这归功于“花瓣”边缘含有不饱和悬空键,有利于CO2的吸附和电荷的转移。另外,当用H2还原步骤处理MoS2纳米花30 min时,CO的生成量比完全未处理或处理时间更短时几乎翻了一番。然而,CVD法需要严格的合成条件要求,并且难以制备原子薄的非层状材料。
非层状结构材料难以通过剥离和刻蚀的方法获得超薄结构。可以采用模板法来制备具有现有2D模板结构的非层状结构材料。例如,可以通过使用K4Nb6O17纳米片作为模板,便捷的水热反应来获得超薄的SnNb2O6纳米片103。AFM分析显示,SnNb2O6的厚度和模板K4Nb6O17一样约为3 nm。与块状SnNb2O6相比,超薄SnNb2O6纳米片具有更大的带隙和更大的负CB电位,电荷转移效率也得到了提高。除了和模板反应之外,在模板上的晶体生长过程也可以用于制备超薄层状材料。Wei的小组通过在CuO纳米板上定向生长氢氧化铁,随后刻蚀掉CuO后加热脱氢合成了独立的半单元晶胞α-Fe2O3纳米片104。显然这一方法可以推广到其他金属氧化物纳米片的制备中。
表面活性剂自组装法被广泛用于制备各种超薄层状材料。它具有工艺简单,产率高的优点。通过表面活性剂十六烷基三甲基溴化铵(CTAB)辅助成功地合成了厚度为0.8 nm的具有[BiO]+-[WO4]2--[BiO]+三明治结构的独立单层Bi2WO6105。由于长碳链CTA+在表面上的阻断作用,层与层的堆积得到了极大的抑制。单层Bi2WO6的[BiO]+-[WO4]2--[BiO]+的三明治亚结构模拟了具有空间电荷的异质结界面,促进了界面上光生载流子的分离。此外,Bi2WO6表面Bi原子的配位不饱和所产生的大量活性位点也是光催化性能优异的原因之一。然而,该方法表面活性剂的选择也是研究的难点并且难以获得均匀的单层纳米片。
此外,谢毅教授等采用一种无机-有机层状杂化中间体方法,合成了各种超薄层状材料,例如Bi2WO6106、In2O3107和Cu2O108。以Bi2WO6为例,油酸根离子最初通过自组装过程与Bi3+相互作用形成层状铋-油酸根络合物前驱体,随后加入Na2WO4后进行水热处理,得到了单层Bi2WO6。CO2吸附等温线和UV/Vis漫反射光谱表明,超大表面积赋予Bi2WO6原子层3倍于普通Bi2WO6的CO2吸附能力和更大的光吸收能力。时间分辨荧光发射光谱表明,单晶胞厚度有助于将载流子寿命从14.7 ns增加到83.2 ns。结果,单层Bi2WO6的甲醇生成速率为75 μmol·g-1·h-1,比以前报道的负载TiO2的沸石和Ag/TiO2高出10倍以上。这种方法本质上是通过有机分子隔离以抑制周期排列的相邻无机碎片之间的相互作用。利用这一有效方法,有望合成各种超薄层状催化剂。
5 提高光催化能力的策略
通过对超薄层状催化剂的厚度调整,掺杂,引入缺陷或复合,光催化性能可能出现明显的提升。文中涉及到的部分催化剂及其改良后的催化性能总结见表1。性能提升的原因可以从光催化原理的角度归因于CO2吸附和催化位点增加,能带结构的调整,电子迁移效率以及电子-空穴分离度增加。这些都将在本节详细讨论。
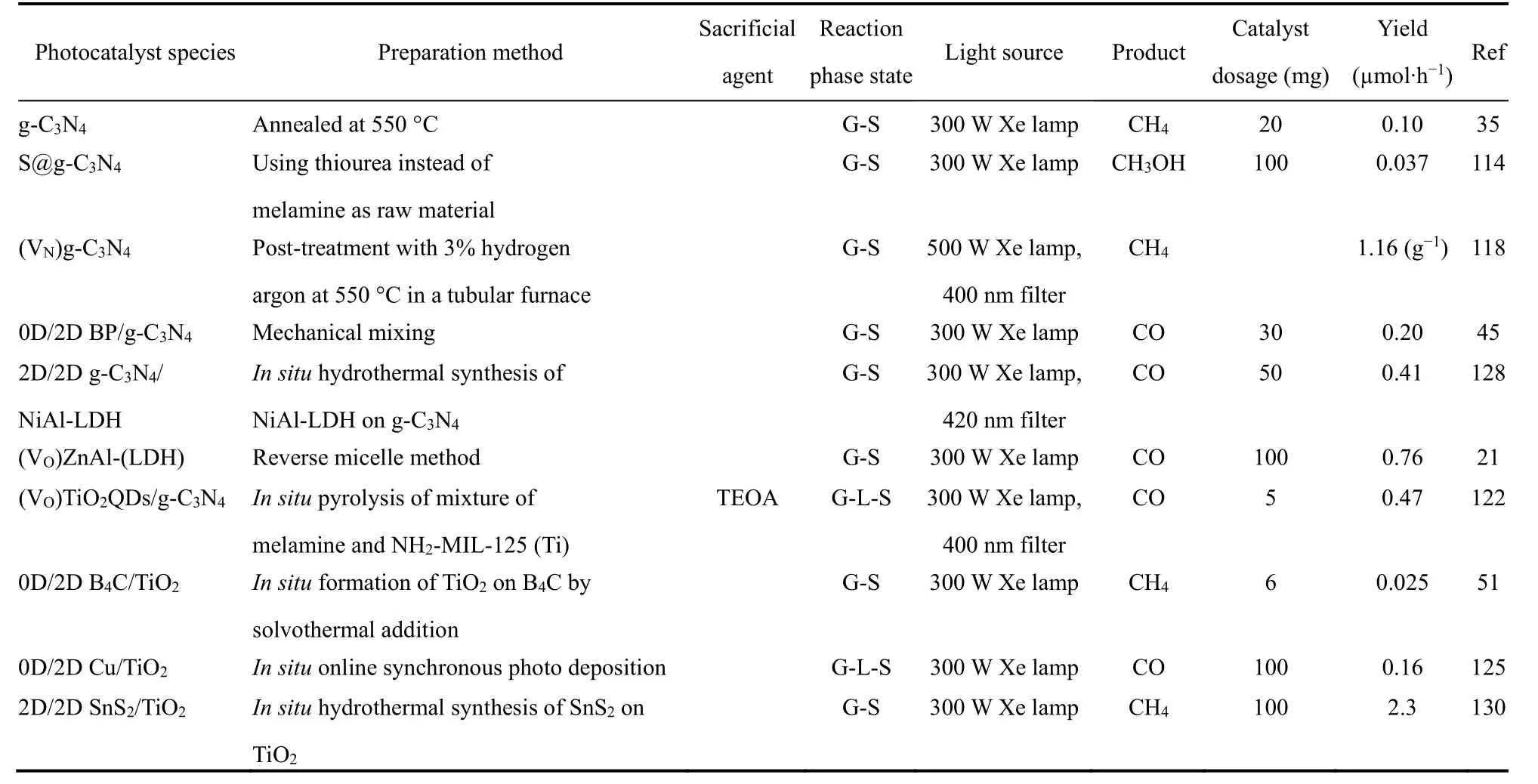
表1 用于光催化CO2还原的超薄层状催化剂的制备和改性方法,光催化活性测试条件和产物产率Table 1 Preparation, modification methods, photocatalytic activity test conditions, and product yield of ultrathin layered catalyst for photocatalytic CO2 reduction
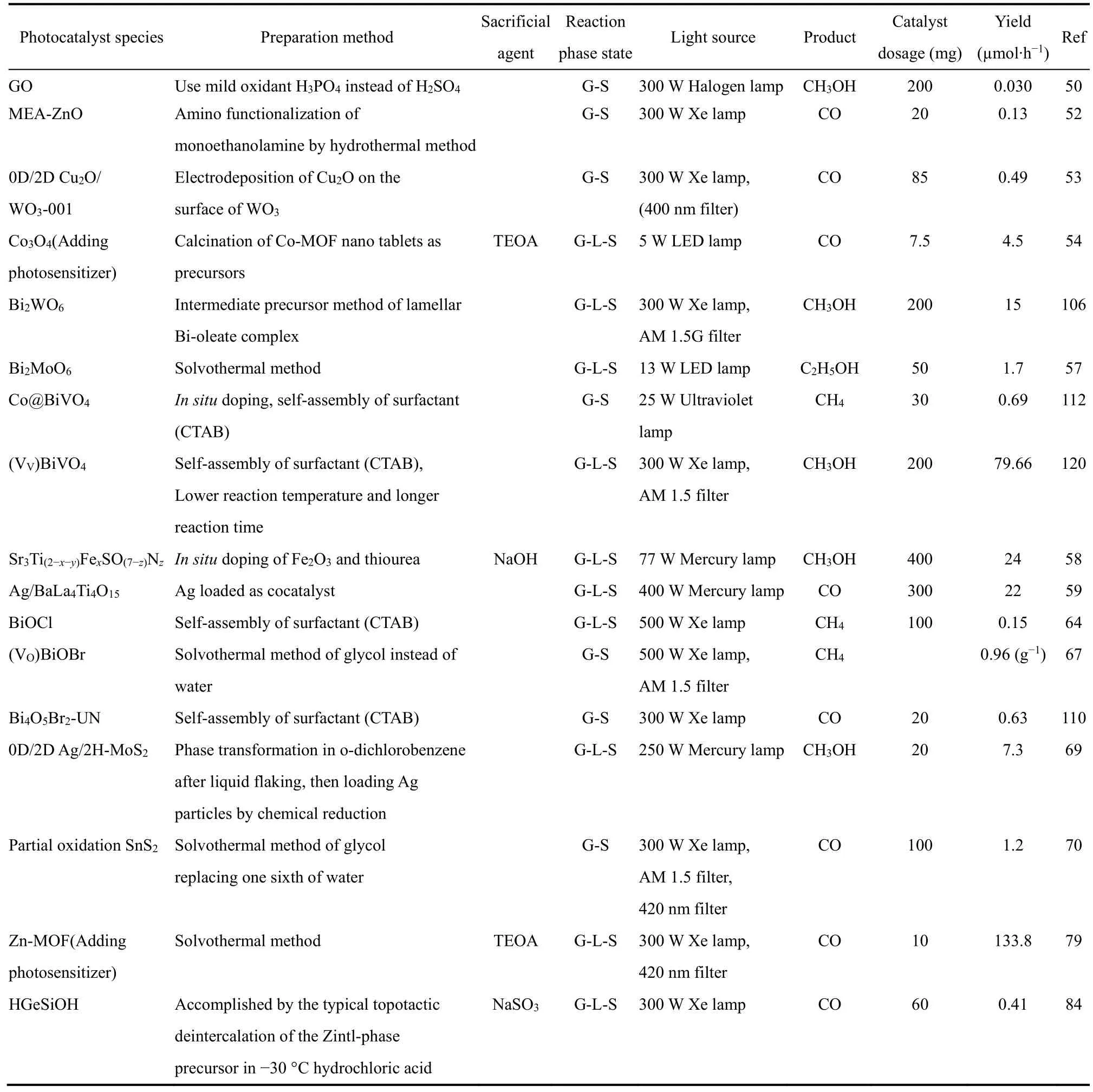
Continued Table 1
5.1 厚度调整
半导体的厚度是影响电子结构调整和光催化性能优化的重要因素。由于众所周知的量子限域效应,当材料厚度逐渐减小时,半导体的带隙将变宽,黑磷就是一个典型的例子。块状BP的实验带隙为0.35 eV,而单层BP的带隙为2.0 eV42,带隙增加高度依赖于层数减少。从态密度(DOS)的计算中可以证实,范德华相互作用在BP及其2D衍生物中随着层数的增加而减小能隙中起着关键作用。层数的增加导致能带在整个布里渊区上分裂,这导致带隙减小109。
另外,当块状材料的厚度减小到单层或很少层时,配位不饱和的表面原子相对于所有原子的比例大大增加。这些表面原子倾向于与其他原子键合以趋向于更稳定。因此,超薄层状结构对CO2的吸附和活化非常有利。相较于厚度6.34 nm的Bi4O5Br2,厚度为2.89 nm的超薄纳米片Bi4O5Br2-UN显示出2倍以上的CO2吸附量,更高的光生载流子分离效率110。此外,厚度的减小导致CB位置变得更负,这赋予了Bi4O5Br2增强的还原能力。在光吸收没有改善的情况下,Bi4O5Br2-UN光催化CO生成速率约为Bi4O5Br2的2.3倍。
5.2 掺杂
掺杂是调整催化剂光催化性能的常用方法之一。进入半导体晶格的异物金属或非金属元素可以改良主体材料的电子结构,以增加光响应范围和促进光生电子的跃迁。但是由于不存在原子渗透通道,通常掺杂只能在块状材料的浅表面上发生。但是对于超薄层状材料,很小的扩散深度即可产生重大影响。因此,掺杂可能是影响超薄层状半导体催化性能的更有效的策略。
5.2.1 金属掺杂
将金属离子掺入半导体晶格可以在其禁带间形成杂质能级,可以导致光吸收边缘的红移,这归因于杂质能级在CB和VB之间的过渡111。另外,光生电子和空穴可以被掺杂剂俘获,增加了局部的电子-空穴分离度。随后,得益于超薄结构,电子被迅速转移到催化剂的表面。将Co掺杂在BiVO4原子层中可以调整其电子结构(图11)112。XPS分析、CO2-TPD和电化学实验表明,Co掺杂使O阴离子周围的电子密度增加,这可能有助于CO2活化和电子转移到活化的CO2。通过密度泛函理论计算的进一步研究表明,Co掺杂会导致在费米能级之上产生新的缺陷能级(图14a,b),这表明较高的空穴浓度有利于水氧化,并且位于价带顶部的Co 3d轨道可能会掺杂充当水氧化的活跃中心。结果,Co掺杂的BiVO4原子层在紫外光照射下实现了23.8 μmol·g-1·h-1的CH4生产率。约为原始BiVO4原子层活性的三倍。
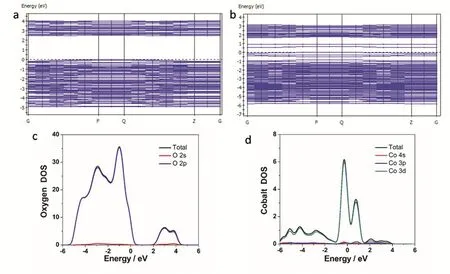
图11 (a) BVO和(b)共掺杂BVO原子层的能带结构;(c, d) BVO原子层的O原子和共掺杂BVO原子层的Co原子的态密度112Fig. 11 Band structure of (a) BVO and (b) Co-doped BVO atomic layers; (c, d) density of states for O atoms of BVO atomic layers and for Co atoms of Co-doped BVO atomic layers 112.
5.2.2 非金属掺杂
对于非金属元素的掺杂,表面掺杂可以在禁带之间建立局部状态作为跳板,有利于激子跃迁。均匀掺杂会导致局部状态与价带的上边缘合并,导致价带上移,带隙减小,从而影响光催化性能113。通过简单地在520 °C下煅烧硫脲来制备掺杂硫的g-C3N4114。从紫外可见光漫反射光谱发现,掺杂硫的g-C3N4(TCN)光吸收边缘可达475 nm,对应的带隙为2.63 eV,这比未掺杂的g-C3N4(MCN)的带隙(2.7 eV)窄。随后利用基于自旋极化密度泛函理论的第一性原理计算进一步研究了TCN和MCN状态的全部和部分态密度,表明TCN和MCN的带隙相同,但TCN样品中存在杂质能级。因此光生电子很容易从TCN样品的杂质能级跃迁到CB或从VB跃迁到杂质能级。进一步用光催化CO2还原法测定样品的光活性,TCN上的CH3OH产率为1.12 μmol·g-1,是MCN的1.5倍。
5.3 缺陷工程
除了掺杂之外,缺陷也会对光催化剂性能产生影响。与缺陷少的同类产品相比,超薄层状光催化剂上的缺陷不仅可以调整电子结构,而且可以提高光吸收能力,表面的局部电荷密度,并且可以促进目标分子的吸附和活化115。得益于原子逃逸能量较小的超薄层状结构,即使缺陷仅在表面产生,也比块状材料上的相对密度大的多。这里介绍两种常见的用于优化超薄层状光催化剂电子结构的缺陷,包括阴离子空位,阳离子空位。
5.3.1 阴离子空位
CO2的吸附是CO2光还原的重要前提。期望找到方法来增加CO2的吸附位并建立强相互作用以促进电子转移以进行有效活化。研究发现,氧空位作为一种和在氧化物材料中的普遍存在阴离子空位,十分有利于CO2吸附116,117。通过受控合成成功制得了厚度分别为2.7和4.1 nm的ZnAl-LDH超薄纳米片和厚度约为210 nm的块状ZnAl-LDH,分别命名为ZnAl-1,ZnAl-2和ZnAl-322。当将ZnAl-LDH的厚度减小至超薄结构时,氧空位(Vo)缺陷的密度将增加,从而将减少相邻Zn离子的配位数,并产生许多配位不饱和的Zn离子。电子自旋共振(ESR)表征显示,在超薄ZnAl-LDH纳米片中构建了Zn+-Vo络合物(图12A-C)。在CO2的光还原过程中,这种配合物可以作为俘获位点来促进CO2的吸附。同时,通过EIS和DFT计算证明,Zn+-Vo配合物还可以用作电子俘获位点,以提高电荷分离效率并促进电子转移至CO2。结果,富含缺陷的超薄ZnAl-LDH纳米片表现出出色的光催化性能,ZnAl-1样品的CO转化效率为7.6 μmol·g-1·h-1。

图12 (A) ZnAl-1,(B) ZnAl-2和(C) ZnAl-3的ESR光谱; (D) (a) ZnAl-1,(b) ZnAl-2和(c) ZnAl-3的紫外可见漫反射光谱22Fig. 12 ESR spectra of (A) ZnAl-1, (B) ZnAl-2, and (C) ZnAl-3; (D) UV-vis diffuse reflectance spectra of(a) ZnAl-1, (b) ZnAl-2, and (c) ZnAl-3 22.
除了氧化物中的氧空位,氮化物上的氮空位118,硫化物上的硫空位119也是常见的阴离子空位,它们都强烈地影响着光催化剂的催化性能。可以通过在还原性气氛下后处理制备氮空位修饰的g-C3N4纳米片(0.326 nm)光催化剂118。XPS定量分析证实了N空位的产生,UV-Vis DRS光谱分析发现空位诱发的中间能隙状态有利于在两步过程中介导多电子激发。这有利于将光吸收扩展到NIR区域以最佳利用太阳能。作为氮空位引入的后效应,延长的荧光衰变寿命充分证明了中间能隙状态在抑制复合速率中的重要作用。结果,存在氮空位的g-C3N4相比于没有空位的g-C3N4的光催化CH4产率提高了3.16倍。以上研究表明,阴离子空位是实现超薄层状光催化剂高效CO2还原的有效改良方法。
5.3.2 阳离子空位
得益于多样的电子构型和轨道,阳离子空位也是优化超薄层状结构电子结构,促进光电荷激发、分离非常有效的途径。例如,可以将钒空位以不同浓度引入到BiVO4纳米片(厚度1.28 nm)中120。通过正电子湮没光谱和X射线荧光(XRF)在原子水平上确认了钒的空位。此外,对于富钒空位的BiVO4纳米片和贫钒空位的BiVO4纳米片,V和Bi的元素比分别确定为0.914和0.976,这进一步表明了钒空位的浓度差异。DFT计算证明,受益于钒空位的存在,可以在BiVO4的带隙中形成新的缺陷能级,使得电子更容易被激发到CB中。另外,可以在VB边缘获得更高的DOS(图13A,B)。因此,钒空位可以增加BiVO4上的光吸收并提高电子电导率。同时,丰富的钒空位使电荷有效分离,可以将载流子的寿命从74.5 ns延长到143.6 ns (图13C)。受益于这些优点,富钒空位的BiVO4纳米片上甲醇生成速率高达398.3 μmol·g-1·h-1,是贫钒空位的BiVO4纳米片上的1.4倍。此外,富钒空位的BiVO4可以持续长达96 h的光还原反应而没有任何明显的光催化活性损失。
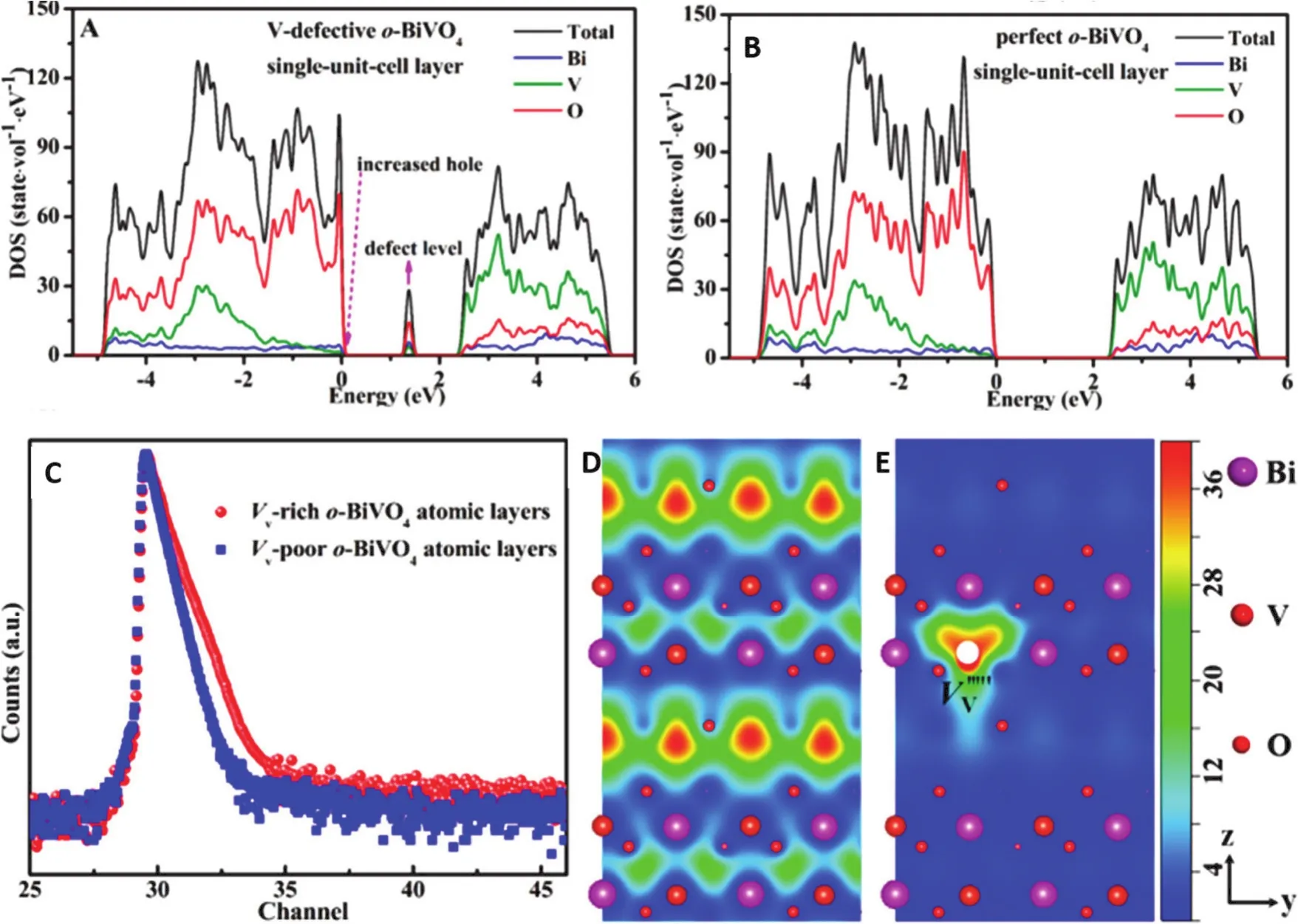
图13 (A)V缺陷o-BiVO4和(B)完美o-BiVO4单晶胞层沿[001]方向的态密度DFT计算;(C)富Vv和贫Vv的o-BiVO4原子层的正电子寿命谱,(D,E)捕获正电子的示意图120Fig. 13 DFT calculations for calculated density of states of (A) V-defective o-BiVO4 single-unit-cell layer slab and(B) perfect o-BiVO4 single-·unit-cell layer slab along the [001] orientation; (C) positron lifetime spectrum of defects characterization for the Vv-rich and Vv-poor o-BiVO4 atomic layers; (D, E) schematic representation of trapped positrons 120.
5.4 复合
得益于超大的表面积,超薄的厚度,光生电荷更加容易扩散到催化剂表面,参与CO2还原反应。因此,在保留超薄层状结构的前提下,引入提高载流子的利用率的表面复合可以大大增强光催化性能。根据另一种材料的结构类型,可分为0D/2D和2D/2D复合。
5.4.1 0D/2D复合
量子点可以接收来自2D材料的光生电荷,以抑制电子-空穴的复合121。因此,将量子点加载到二维纳米片上构建0D/2D异质结构是促进电荷分离的有效途径。通过将NH2-MIL-125 (Ti)和三聚氰胺原位热解可以合成富氧空位TiO2量子点/g-C3N4纳米片异质结构122。飞秒和纳秒泵浦探测瞬态吸收光谱揭示了TiO2-x/MCN中载流子的快速衰减是由于电荷通过异质结界面从2D-g-C3N4转移到0DTiO2。电子转移发生在超快的亚皮秒时间尺度上,随后伴随着由浅俘获位点介导的复合弛豫。快速电荷转移极大地促进了电荷分离和抑制光生电荷复合。另外,所获得的TiO2-x/MCN材料具有增强的可见光吸收,优异的CO2吸附,大的表面积的优点,从而显着增强了光催化CO2性能。
为了进一步提高光催化活性,将0D纳米粒子的尺寸从量子点缩小到单原子(分子)也是可行的策略。单原子催化剂由分散或配位形式存在于载体表面上的单个原子组成,自张涛院士和他的同事首次提出单原子催化以来,它已经成为催化领域一个非常热门的项目123,124。由于单原子具有高活性,它与2D载体之间的化学键将变得更强,并且电荷转移过程将变得更加容易。例如,Jiang等125采用原位在线同步光沉积法把铜原子负载到TiO2纳米片上,随后用氙灯进行光催化实验,CO产率达到了原始TiO2纳米片的10倍。紫外-可见吸收光谱显示,Cu原子的负载增加了催化剂的可见光吸收(图14c)。将催化剂在空气中暴露24 h再洗涤干燥命名Cu/TiO2-2/24 h。高分辨率O 1s XPS结果显示,TiO2纳米片上高度分散的Cu0易于氧化(图14d)。Cu 2p XPS结果则显示,Cu氧化后易从TiO2中脱离(图14b)。
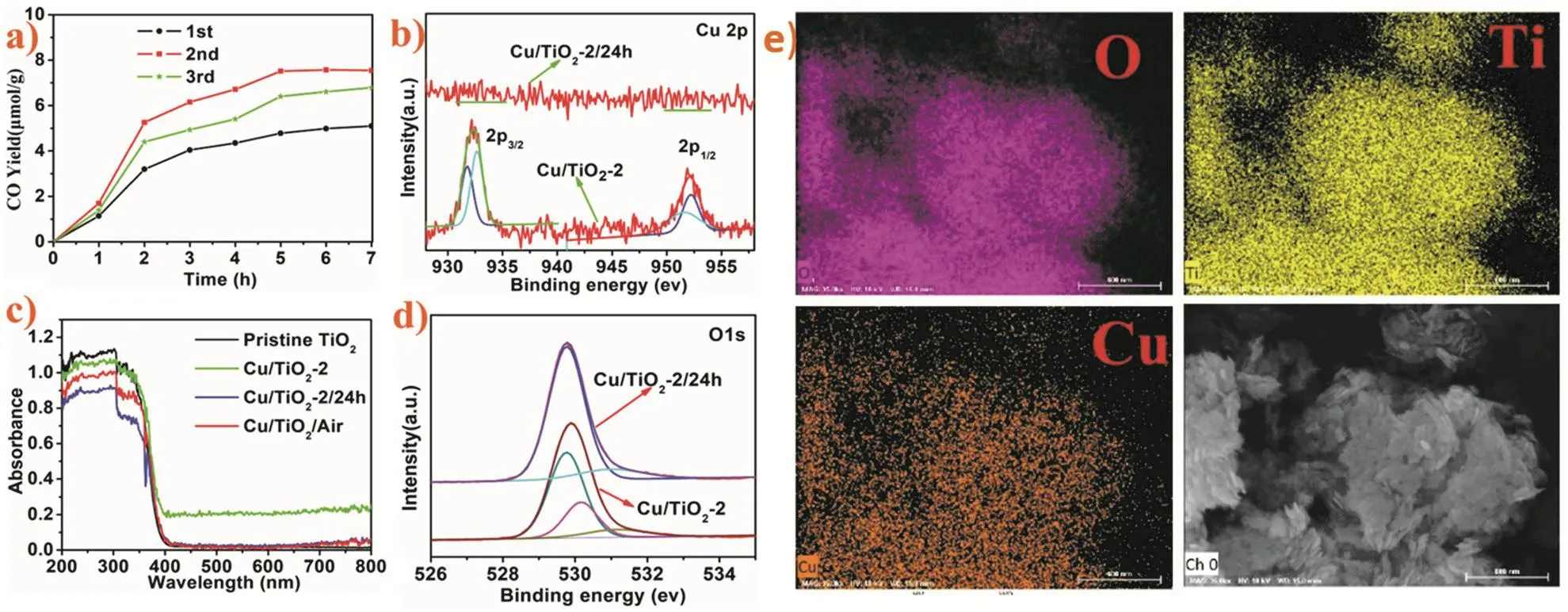
图14 (a) Cu/TiO2-2三次连续运行的光催化CO2 → CO还原活性,(b) Cu/TiO2-2/24 h和Cu/TiO2-2的Cu 2p区的高分辨率XPS光谱,(c) 原始TiO2,Cu/TiO2-2,Cu/TiO2-2/24 h和Cu/TiO2/空气的紫外可见吸收光谱,(d) Cu/TiO2-2/24 h和Cu/TiO2-2 O 1s区域的高分辨率XPS光谱,(e) Cu/TiO2-2的EDS映射图像125Fig. 14 (a) Photocatalytic CO2 → CO reduction activity of Cu/TiO2-2 for three consecutive runs, (b) high-resolution XPS spectra of the Cu 2p region of Cu/TiO2-2/24 h and Cu/TiO2-2, (c) UV-Vis absorption spectra of pristine TiO2,Cu/TiO2-2, Cu/TiO2-2/24 h, and Cu/TiO2/Air, (d) high-resolution XPS spectra of the O 1s region of Cu/TiO2-2/24 h and Cu/TiO2-2, (e) EDS mapping images of Cu/TiO2-2 125.
5.4.2 2D/2D复合
对于超薄层状材料,2D/2D复合是增强光催化性能最有效的策略之一126。首先,2D/2D复合可使层状催化剂形成较大的亲密界面,这对于电子空穴对的形成和光催化剂的稳定性是有利的;其次,异质结的形成促进2D/2D半导体之间电子-空穴对的分离和传输;最后,两种半导体协同作用导致光吸收范围扩大,从而提高了对太阳光的利用127。例如,可以用原位水热法合成g-C3N4/NiAl-LDH 2D/2D异质结催化剂128。带正电的NiAl-LDH板和带负电的g-C3N4纳米板组装在一起建立了强界面体系(图15d)。所制备的g-C3N4/NiAl-LDH在可见光照射下具有良好的光催化CO2还原性能,CO产生速率为8.2 μmol·g-1·h-1,是纯g-C3N4的5倍,是纯NiAl-LDH的9倍。高性能主要归功于g-C3N4与NiAl-LDH之间的协同效应,包括2D/2D组装产生的强界面接触、增加的电子-空穴分离性能和激子的转移效率提升(图15a,b)。更出色的是,活性在连续的实验运行之后没有任何明显的变化,异质结仍然具有高度的光稳定性。
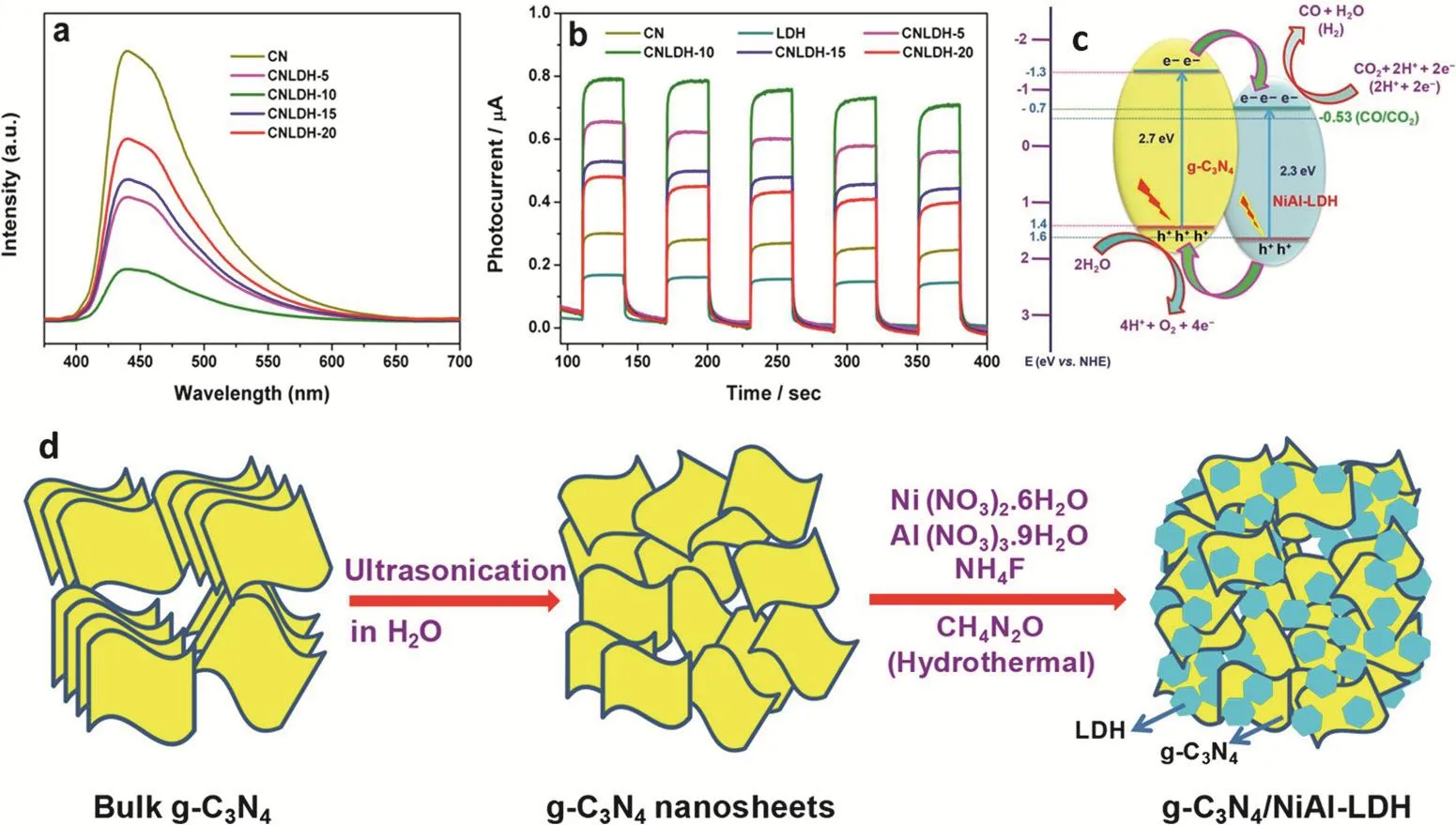
图15 (a) g-C3N4,NiAl-LDH和g-C3N4/NiAl-LDH异质结光催化剂的PL光谱和(b)瞬态光电流响应;(c) g-C3N4/NiAl-LDH异质结中可能的CO2光还原机理的示意图;(d) g-C3N4/NiAl-LDH杂化异质结的合成过程示意图128Fig. 15 (a) PL spectra and (b) transient photocurrent responses of g-C3N4, NiAl-LDH, and g-C3N4/NiAl-LDH heterojunction photocatalysts; (c) Schematic illustration of the proposed mechanism for CO2 photoreduction in the g-C3N4/NiAl-LDH heterojunctions; (d) schematic illustration of the synthesis process of g-C3N4/NiAl-LDH hybrid heterojunctions 128.
除了g-C3N4/NiAl-LDH之外,还有非常多关于2D/2D复合以提升光催化性能的报道129。例如,She等以高能(001)和低能(101)平面2D-TiO2纳米片为光吸收半导体,2D-SnS2纳米片为助催化剂,采用水热法合成了2D-2D-SnS2/TiO2复合光催化剂130。结果表明,合成的2D-2D SnS2(5%)/TiO2样品经300 W氙灯照射1 h后,甲烷产率可达23 μmol·g-1·h-1,是P25的20倍。电荷转移遵循Z方案转移模式。得益于2D SnS2与2D TiO2之间的紧密界面接触,光诱导载流子的分离度增加,同时氧化还原电位增加,这是光催化性能提升的主要原因。
6 结论与观点
光催化CO2还原是一项非常有前景的CO2利用途径,它可以利用取之不尽的太阳能将CO2转化为高附加值化学品。在开发光催化性能优于块状材料的超薄层状材料方面,目前已经取得了令人瞩目的成果。许多本是块状的催化剂被制成纳米片(如TiO2,ZnO),另外也有新的超薄层状材料被开发(如HGeSiOH)。各种不同的改良方法几乎都已经被尝试来提升已被开发的2D催化剂的性能,如掺杂,减少厚度,构造缺陷,复合等。开发与改良超薄层状催化剂是光催化CO2还原领域一个充满希望与前途的方向。
尽管超薄层状光催化剂的研究已经取得了长足进步,但在CO2还原领域它仍然面临着大量挑战。首先,厚度可控的超薄层状半导体的大规模制备方法目前仍是一个难题。催化剂的大规模、低成本、性能可靠的生产方法对于潜在的工业应用非常重要,因此应关注超薄层状材料生产方法的改进。其次,超薄结构给催化剂带来了高表面积的同时,表面易被光生空穴和电子分解,光稳定性相较于晶体结构的下降也是一个必需解决的问题。再次,光催化CO2还原最理想的产物是CO,不仅产率高,而且无法低成本地在自然界中大量获得。但目前光催化CO2还原研究主要集中在催化活性的提升的方向,模拟计算也大多停留在半导体本身性质和CO2吸附阶段,对CO2转化的机理和产物选择性的研究还很少。超薄层状结构提供了更简单、理想的模型来研究光催化机理,理论计算与先进的原位表征的协同利用有助于对CO2转化的微观过程的了解,未来的最终目标是定制催化剂的结构和带隙来制备高转化率、高目标产物选择性的催化剂。最后,由于目前开发的每种光催化剂都有各自的弱点,仍没有一种光催化剂同时满足低成本,高稳定性,高CO2还原活性这三个要求。目前获得最佳性能的理想方法是不同材料的复合以产生协同效果。因此在研究现有光催化剂改良方法的同时,仍需要付出巨大的努力来开发新的性能更加出色的光催化剂。
猜你喜欢
物理学报(2022年17期)2022-09-14
陶瓷学报(2021年5期)2021-11-22
学苑创造·A版(2020年9期)2020-10-13
陶瓷学报(2019年6期)2019-10-27
重型机械(2019年3期)2019-08-27
陶瓷学报(2019年5期)2019-01-12
浙江农业科学(2016年11期)2016-05-04
新疆钢铁(2015年2期)2015-11-07
中国塑料(2015年10期)2015-10-14
读者欣赏(2014年6期)2014-07-03
