
含三齿席夫碱配体锇(Ⅵ)的氮合物与CN-的反应活性研究
2021-06-01 08:36:00向景周鑫王礼鑫张旭罗丽娟
长江大学学报(自科版) 2021年3期
向景,周鑫,王礼鑫,张旭,罗丽娟
长江大学化学与环境工程学院,湖北 荆州 434023
近年来,金属氮合物由于其独特的电化学性质和光学性能受到了人们广泛的关注[1-8],探索金属氮合物的新反应活性具有较为重要的基础研究意义。在生物和化学体系中,金属氮合物是催化氮气转化为氨的关键中间体。除了在氮气固定中起重要作用外,金属氮合物还可以用作多种有机底物的氮化试剂[9-11]。近二十年来,相关课题组利用含多联吡啶锇(Ⅵ)氮合物与各种亲核试剂反应生成了许多新颖的N- -C、N- -N、N- -P和N- -E (E=O, S, Se)成键反应[12-18]。在这些金属氮合物中,以刘大铸课题组的研究最为突出,他们合成了具有高亲电活性的含席夫碱钌(Ⅵ)氮合物[RuⅥ(N)(L)(CH3OH)]+(L=N,N’-双(水杨酸)-o-环己基二胺阴离子)[19-21]。该RuⅥN配合物在室温条件下就能够与各种稳定的有机底物反应,其中最具有代表性的反应活性就是它能够与烯烃发生反应生成含三元环的氮杂环化合物和活化各种稳定的烷烃的C- -H键[22-29]。
最近几年,笔者也研究了金属锇的氮合物[OsⅥN(sap)(Cl)(OH2)](H2sap= N-水杨酸-o-氨基苯酚)在不同溶剂中和不同比例的CN-的反应[30]。该反应最初发生在三齿席夫碱配体上,而Os≡ ≡N基团保持不变。当反应在甲醇溶剂中进行时,得到了一种有趣的顺磁性氢氰氨基配合物(PPh4)2[OsⅢ{N(H)CN}(O^N)(CN)3]) (O^N = 2-(2-羟基苯基)苯并恶唑)。该氢氰氨基配合物既能表现出亲电性又能表现出亲核性。但更重要的是,它可以作为制备各种胍配合物的合成子[31-33]。而这些含胍基配合物又可以进一步转化为各种具有独特LMCT特性的发光锇的氮合物[34]。研究表明,这些新型发光的锇的氮合物在激发态下具有较高的活性,这明显的不同于它们在基态条件下的反应性[35,36]。
为进一步制备发光的锇的氮合物,笔者合成了一个新的含三齿席夫碱配体的配合物mer-[OsⅥ(L1)(N)(Cl)(H2O)] (1) (H2L1= 1-((2-羟基苯基)亚胺)甲基)萘酚-2-醇),并研究了它和不同比例的氰根在乙腈中的反应。分别得到了2个新的锇的氮合物:fac-(PPh4)[OsⅥ(N)(L1)(CN)2] (2)、fac-(PPh4)2[OsⅥ(N)(L2)(CN)2] (3) (H3L2=2-(2-羟基萘-1-基)-2-((2-羟基苯基)氨基)乙腈)。当在水中进行相同的反应时,发现配合物(1)易于分解,并分离出产率较低的锇(Ⅵ)的氮合物(PPh4)2[Os(L3)(CN)4] (4)(L3=6-亚氨基环己烷-2,4-二烯-1-酮)(见图1) 。
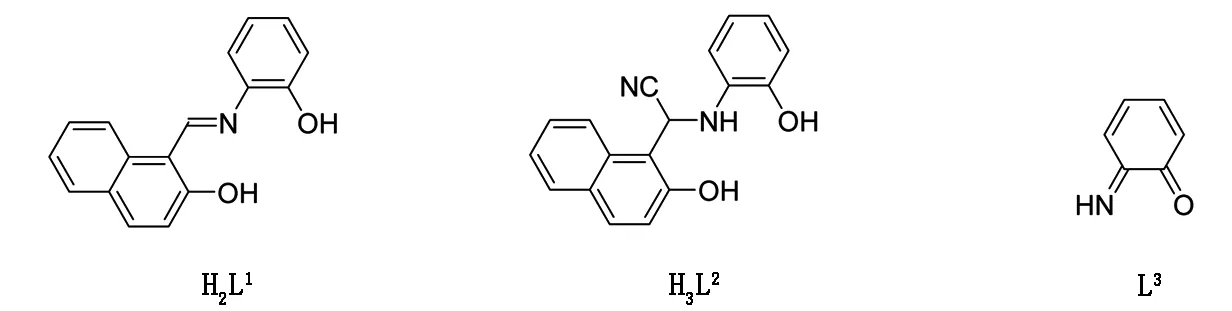
图1 配体H2L1、H3L2和L3的结构Fig.1 The structures of ligands H2L1, H3L2 and L3
1 试验部分
1.1 试剂与仪器
1)试剂。用于电化学测试的[nBu4N]PF6(Aldrich)在沸腾的乙醇中重结晶三次,并在120℃真空下干燥24h。用于电化学测试的乙腈(Aldrich)使用氢氧化钙蒸馏干燥。其他化学试剂均为试剂级,使用时无需进一步纯化。除另有说明外,所有操作均未采取预防措施以排除空气或湿气。
2)仪器。Nicolet 360 型傅里叶红外光谱仪(KBr压片),Vario EL型元素分析仪,PE-SCIEX API 365 三重四极杆质谱仪,PAR模型273恒电位器(玻璃碳为工作电极、Ag/AgNO3(0.1mol/L的乙腈溶液)为参比电极和以二茂铁(Cp2Fe)为内标的铂丝为对电极)。
1.2 试验方法
1.2.1 X-射线晶体学测试
在牛津CCD衍射仪上使用石墨单色化的MoKa射线 (λ= 0.71073Å) 为光源对配合物的晶体结构进行测试。采用多次扫描法进行吸收校正。晶体结构由重原子Patterson方法或直接法解析,利用SHELX-97通过全矩阵最小二乘法进行细化,并使用傅里叶技术进行扩展[37,38]。所有非氢原子都是各向异性的。氢原子是由SHELXL-97产生的。根据骑乘模式计算氢原子的位置,热力学参数为伴生C原子的1.2倍,并参与最终残差因子R的计算。所有计算均使用teXsan晶体学软件进行[39,40]。
1.2.2 合成与表征
1)配合物(1):mer-[Os(N)(L1)(Cl)(H2O)] 。将[nBu4N][OsⅥ(N)(Cl)4] (588mg,1.0mmol)溶于30mL甲醇中,加入1倍的配体H2L1(263mg, 1.0mmol),然后把混合液体加热回流1d。将生成的橙红色沉淀过滤,用1mL的甲醇清洗,室温晾干。产率:65%。红外光谱(KBr, cm-1):ν(Os≡ ≡N) 1098。元素分析C17H13ClN2O3Os理论值(w/%):C(39.34),H(2.52),N(5.40); 实验值(w/%):C(39.25),H(2.80),N(5.31)。核磁共振氢谱 (400MHz, CDCl3,ppm):δ9.82(s,1H;CH= =N ), 8.26~8.29(d,1H,J=8.4Hz, Ar-H),8.12~8.14 (d,1H,J=9.2Hz,Ar-H),7.96~7.88 (m,1H,Ar-H),7.75~7.81(dd,2H,J=16.6,8.2Hz;Ar-H),7.57~7.61 (t,1H,J= 7.4Hz;Ar-H),7.51~7.53 (d,1H,J=7.1Hz;Ar-H),7.33~7.37 (t,1H,J= 7.8Hz;Ar-H),7.05~7.09 (t,1H,J= 7.8Hz;Ar-H)。
2)配合物(2):fac-(PPh4)[OsⅥ(N)(L1)(CN)2]。将2倍的Et4NCN (63mg,0.4mmol) 加入到15mL含有配合物(1) (104mg, 0.2mmol) 的乙腈溶液中,然后把混合溶液在室温下搅拌1d,再加入PPh4Cl (75mg,0.2mmol)。将溶剂浓缩至约2mL,加入乙醚(5mL)生成橙红色沉淀,过滤,用1mL乙醚进行洗涤,室温晾干。产率:78%。将乙醚缓慢扩散到乙腈溶液中,得到了适合X-射线晶体学测试的晶体。红外光谱(KBr, cm-1):ν(C≡ ≡N) 2146,2138;ν(Os≡ ≡N) 1055。电喷雾-质谱:m/z519 (M-)。元素分析C43H31N4O2OsP理论值(w/%):C(60.27),H(3.65),N(6.54)。实验值(w/%):C(60.20),H(3.72),N(6.52)。核磁共振氢谱(400MHz,CD3CN,ppm):δ8.72(s,1H;CH= =N ),8.10~8.12(d,1H,J=8.2Hz;Ar-H),7.91~7.95 (td,4H,J=7.4,1.8Hz;Ar-H), 7.84~7.88(m,2H;Ar-H),7.79~7.81(d,1H,J=8.0Hz;Ar-H),7.66~7.78(m,16H;Ar-H),7.63~7.65(d,1H,J=8.2Hz;Ar-H),7.41~7.45 (m,1H;Ar-H),7.17~7.23 (m,1H;Ar-H),6.89~6.86 (m,2H;Ar-H),6.77~6.83 (m,1H;Ar-H)。
3)配合物(3):fac-(PPh4)2[OsⅥ(N)(L2)(CN)2]。将4倍的Et4NCN (126mg, 0.8mmol) 加入到15mL含有配合物(1) (104mg,0.2mmol) 的乙腈溶液中,然后把混合溶液在室温下搅拌1d,再加入PPh4Cl (75mg, 0.2mmol)。将溶剂浓缩至约2mL,加入乙醚(5mL)生成橙红色沉淀,过滤,用1mL乙醚进行洗涤,室温晾干。产率:65%。将乙醚缓慢扩散乙腈溶液中,得到了适合X-射线晶体学测试的晶体。红外光谱(KBr, cm-1):ν(C≡ ≡N), 2246, 2146, 2138;ν(Os≡ ≡N)1035。电喷雾-质谱:m/z519[(M-CN)-];546[(M+H)-]; 884[(M+PPh4)-]。元素分析:C68H51N5O2OsP2理论值(w/%):C(66.82); H(4.21); N(5.73)。实验值(w/%):C(66.78); H(4.24); N(5.71)。核磁共振氢谱(400MHz, CD3CN,ppm):δ7.89~7.93 (ddt, 8H,J=2.9, 2.1, 1.3Hz; Ar-H), 7.83~7.85 (d,1H,J= 8.7Hz; Ar-H), 7.63~7.78 (m, 32H; Ar-H), 7.50~7.52 (d,1H,J= 7.2 Hz; Ar-H), 7.37~7.41 (ddd,1H,J=8.4, 6.9, 1.4Hz; Ar-H), 7.24~7.26 (d,1H,J= 9.0Hz; Ar-H), 7.01~7.05 (t,1H,J= 7.4Hz; Ar-H ), 6.92 (s,1H; C- -H(CN)- -N), 6.70~6.74 (m,1H; Ar-H), 6.51~6.55 (m,1H; Ar-H), 6.47~6.49 (d,1H;J= 9.0Hz; Ar-H), 6.23~6.31 (m, 2H; Ar-H)。
2 结构与讨论
2.1 配合物(1)的合成、表征及其与CN-的反应
与笔者先前报道的配合物[OsⅥ(sap)(N)(Cl)(H2O)][30]相似,将[nBu4N][OsⅥ(N)Cl4]和适量的H2L1在甲醇中回流就可以制备出配合物(1)(产率约为65%)。配合物(1)在空气中较为稳定,通过分离得到橙红色的微晶样品。配合物(1)并进一步通过核磁共振氢谱、红外光谱和紫外可见吸收光谱和元素分析进行了表征。配合物(1)具有抗磁性的低自旋d2-电子组态,这可以通过在CDCl3的核磁共振氢谱中,在正常范围内所展现出的尖峰得以证明。在低磁场δ9.82ppm处的单峰信号被指认为配体L1中的亚胺基团的质子信号(见图2),这一单峰信号与含四齿席夫碱配体的锇(Ⅵ)的氮合物[OsⅥ(N)(salophen)Cl] (salophen=N,N’-双(水杨酸)-邻苯二胺二阴离子)[41]的亚胺基团质子的化学位移(δ9.87ppm)相一致。在配合物(1)的红外光谱中,在1098cm-1处展现出一个中等强度的峰,这是相关锇(Ⅵ)的氮合物的ν(Os≡ ≡N)的特征振动峰。
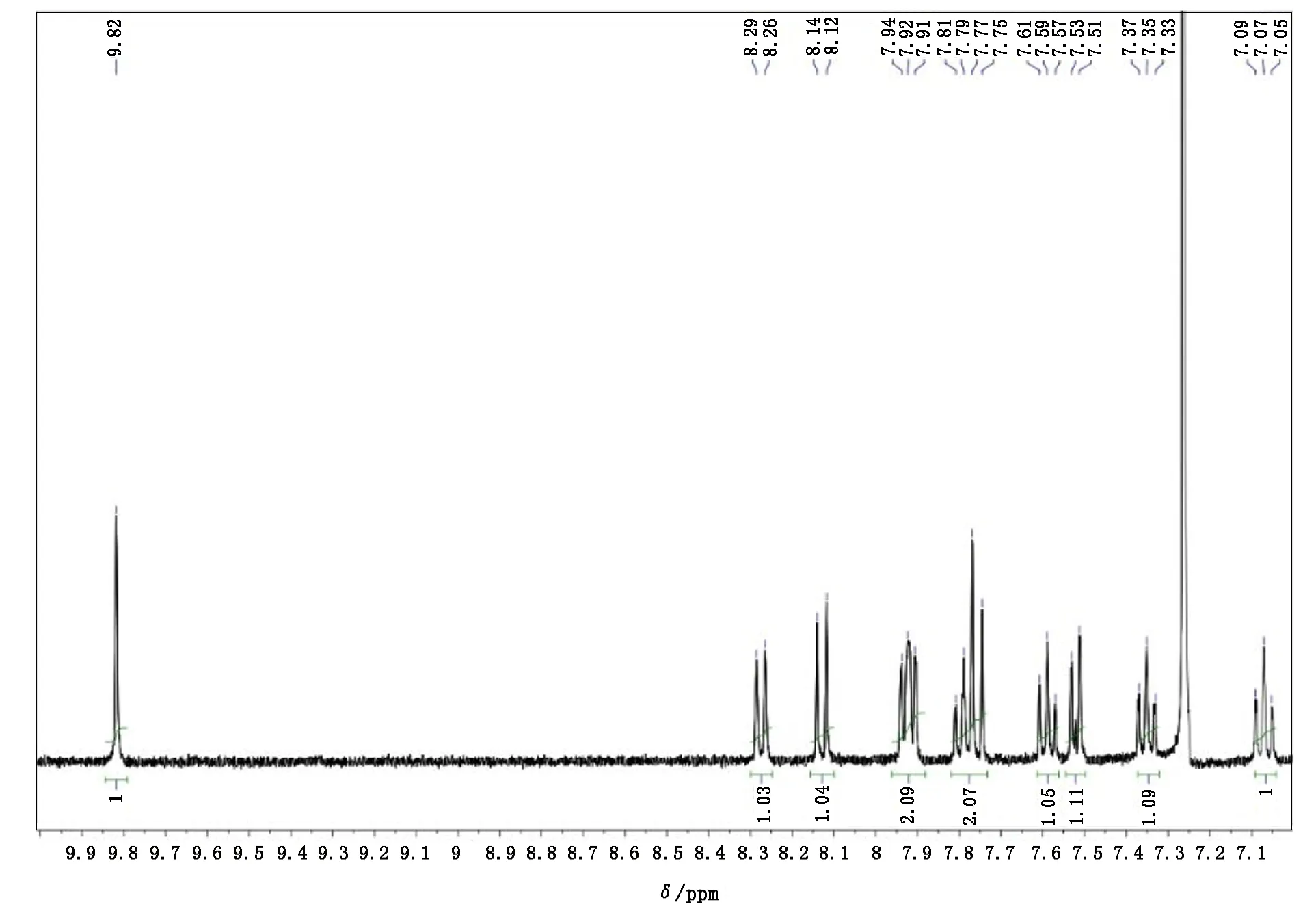
图2 配合物(1)的核磁共振氢谱(400MHz, CDCl3)Fig.2 H NMR of the complex (1) (400MHz, CDCl3)

图3 配合物(1)和CN-在不同溶剂中的反应Fig.3 The reactions of the complex (1) with CN- in different solvents


注:插图(a)、(b)分别为m/z 519的实验和模拟同位素分布图。图4 配合物(2)的电喷雾质谱Fig.4 ESI-MS of the complex (2)

图5 配合物(2)的核磁共振氢谱 (400MHz, CD3CN)Fig.5 H NMR of the complex (2) (400MHz, CD3CN)

图7 配合物(3)的核磁共振氢谱 (400MHz, CD3CN)Fig.7 H NMR of the complex (3) (400MHz, CD3CN)
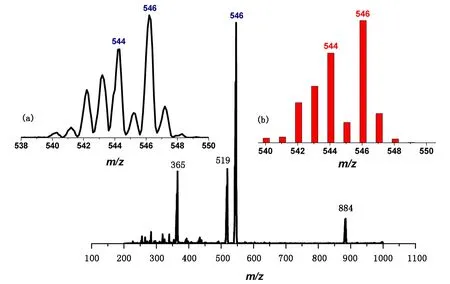
注:插图(a)、(b)分别为m/z 546的实验和模拟同位素分布图。图6 配合物(3)的电喷雾质谱Fig.6 ESI-MS of the complex (3)
2.2 配合物(2)的晶体结构
配合物(2)结晶于单斜晶系,P21/n空间群。阴离子结构的ORTEP图如图9所示,表1和表2是配合物(2)部分的键长和键角数据。金属锇中心由1个fac-L1、2个CN-和1个配位氮原子构成一个六配位的扭曲的八面体构型。与之前报道过的配合物[OsⅥ(sap)(N)(Cl)(H2O)][30]相似,Os- -N1的键长(1.654(3) Å) 比其他相关的配合物的键长要更长[12-18],但是和[OsⅥ(N)(NO2- -Q)2Cl][42]中的键长相一致。由于配位氮原子的反式效应影响,Os1原子偏离由 C1、C2、O1和 N4原子组成的赤道面位置0.245Å。Os- -O2 (2.1516(18) Å)的键长比Os- -O1 (2.032 (2) Å)的键长0.12 Å。2个Os- -C键的键长则非常接近,分别为2.078(3)Å和 2.045(3) Å。配体L1是非共面的,可以分为羟基苯酚和萘酚2部分,其二面夹角为33.4°。N4- -C9的键长为1.300(3) Å,这与亚胺基团的双键特性一致。通过对堆积图的观察发现,碳与苯环质心的距离为3.508 Å,说明存在边界-面的π- -π堆积作用。
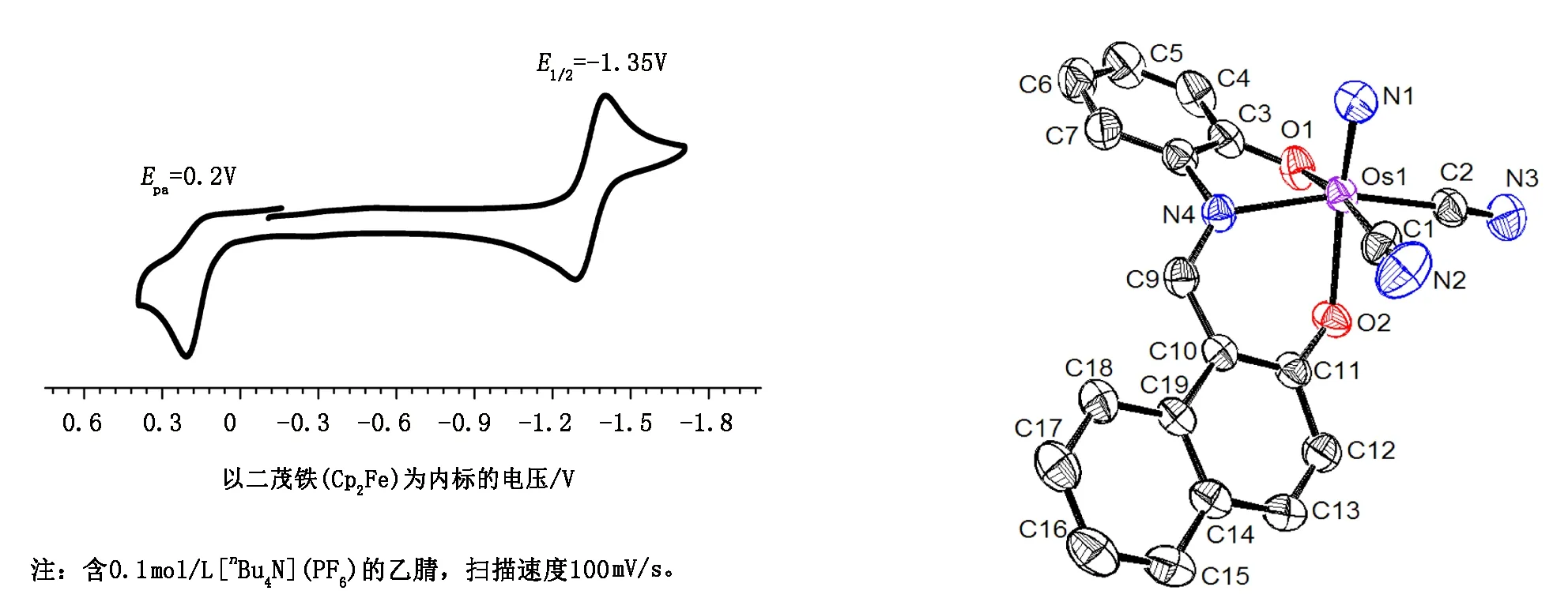
图8 配合物(4)的循环伏安法图谱 图9 配合物(2)阴离子结构示意图 Fig. 8 CV of the complex (4) The anionic structure of the complex (2)

表1 配合物(2)的键长
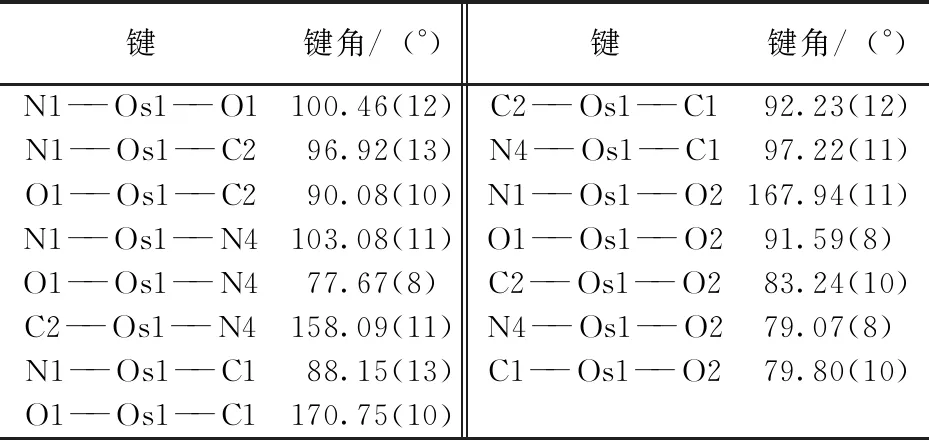
表2 配合物(2)的键角
2.3 配合物(3)的晶体结构
配合物(3)结晶于三斜晶系,P-1空间群。图10所展示的是阴离子结构图,并标记了部分原子的编号及选择的键参数见表3和表4。与配合物(2)相似,金属锇中心也是一个扭曲的八面体构型。Os- -N4的键长为 1.658(2) Å,这和配合物(2)中Os- -N的键长相似,也符合其三重态键的特征。Os1原子位于由 C1、C2、N5和 O1构成的赤道面外,距离为0.275 Å。两个芳香环形成的二面夹角为99.3°,这与配合物(2)中的二面夹角相比要大一点。这也表明,CN-亲核加成到亚胺键形成新的C- -N单键后,空间位阻效应减弱。Os1- -O2的键长(2.146(1) Å)远长于Os1- -O1的键长(2.021(1) Å),这再次说明配位氮原子的反式效应较强。Os1- -N5的键长(2.009(2) Å)比Os- -N (亚胺)短,说明N5是氨基氮。根据单键性质,C10- -N5和C10- -C3的键长分别为1.449(3)和1.486(3) Å。N5- -C10- -C3和N5- -C10- -C11的键角分别为111.7(2)°和115.9(2) °,表明C10原子是sp3杂化的。

图10 采用ORTEP绘制的配合物(3)阴离子结构示意图 Fig.10 The anionic structure of the complex (3) drawn by ORTEP

表3 配合物(3)的键长

表4 配合物(3)的键角
2.4 配合物(4)的晶体结构
配合物(4)结晶于正交晶系,Pnma空间群。阴离子结构的ORTEP图如图11所示,表5和表6为配合物(4)部分的键长和键角数据。在配合物(4)中,金属锇中心也是由一个双齿配体L3和四个CN-配体构成的6配位的扭曲的八面体几何构型。有一个镜像平面穿过L3平面。Os1- -C键长在1.952(8) ~ 2.078(6) Å范围内,轴向的C5- -Os1- -C5i键角为162.0(2)°,接近直线角度。Os1- -N4和Os1- -O1的键长分别为1.979(5) Å和2.115(4) Å。C4- -N4和C3- -O1键长分别为1.365 Å和1.291Å,这是典型的C= =N和C= =O双键的特征。晶体结构表明,2-氨基苯酚被氧化为中性的6-亚氨基环己烷-2,4-二烯-1-酮。
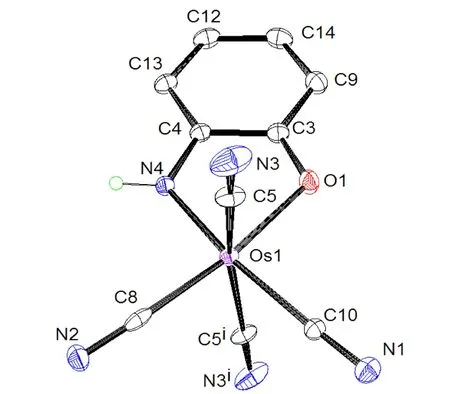
图11 配合物(4)阴离子结构示意图Fig.11 The anionic structure of the complex (4)

表5 配合物(4)的键长

表6 配合物(4)的键角
3 结论
通过对含席夫碱配体的OsN与CN-的反应活性研究,得出如下结论:
1)制备了具有三齿席夫碱配体锇(Ⅵ)的氮合物mer-[OsⅥ(L1)(N)(Cl)(H2O)](1),并研究了配合物(1)与不同比例的CN-在不同溶剂中的反应。
2)当一个配位水分子和一个氯原子被两个CN-配体取代时,席夫碱配体的构型由经式变为面式。当加入更多的CN-时,可能导致CN-亲核加到fac-L1的亚胺键上,而OsN基团则保持不变。
3)在H2O中加入过量的CN-很容易导致配合物(1)的分解,氮原子很可能经历两分子的Os≡ ≡N的耦合来释放N2。在这些反应中没有观察到类似的配体向双齿O^N配体的转化,只能分离出非常少的四氰基锇(Ⅱ)产物。
猜你喜欢
中学化学(2024年3期)2024-06-30 15:19:19
云南化工(2023年7期)2023-08-01 07:59:34
现代临床医学(2022年4期)2022-09-29 07:36:10
高中数理化(2022年16期)2022-09-14 13:57:06
中国抗生素杂志(2022年7期)2022-08-18 03:22:36
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:38
长春师范大学学报(2019年4期)2019-04-29 05:51:36
电源技术(2015年12期)2015-08-21 08:58:30
邵阳学院学报(自然科学版)(2015年2期)2015-06-05 12:22:39
