
Cr基催化剂的丙烷脱氢活性位结构及反应机理的研究进展
2021-05-28 02:01解则安
沈阳师范大学学报(自然科学版) 2021年2期
解则安, 曹 奇, 曾 敬, 张 霄, 赵 进, 赵 震
(沈阳师范大学 化学化工学院, 沈阳 110034)
0 引 言
随着全球石油资源的逐渐减少以及国际油价的大幅波动,烯烃工业正在不断寻找大量新的易得原材料和更有效的生产转化方式来实现增产。利用天然气、页岩气等丰富廉价的资源中的低碳烷烃脱氢制烯烃具有巨大的经济价值。目前工业上正在大力投入和发展的丙烷脱氢工艺,具有反应过程简单、清洁无污染等特点,取得了较好的经济社会效益。
当前丙烷脱氢工艺在工业中已得到了实际应用,UOP公司开发的Olefelex与Lummus-Houdry 开发的 Catolefin 工艺是目前综合技术成熟度最高、装置建设经验最丰富的2套 PDH工艺,在工业界得到了最为广泛的应用,其所用催化剂分别是PtSn/Al2O3体系和Cr2O3/Al2O3体系。Pt基催化剂活性高、丙烯选择性好,催化剂本身毒性小,但其在高温条件下积碳严重,稳定性差,而且生产成本高,对原料纯度要求高。Cr2O3基催化剂具有活性高、价格低、对原料杂质要求低等优点,但催化剂容易积碳失活而需要频繁的再生,大大降低了设备的生产能力。因此,制备高初活性同时长寿命的廉价丙烷脱氢催化剂是目前研究的热点[1-2]。
Cr基催化剂用于烷烃脱氢已经发展了近90年,Jesper等[3]和Marktus等[4]关于烷烃脱氢的综述中,分别详细总结了应用于丙烷无氧脱氢和有氧脱氢(CO2弱氧化剂辅助)的Cr基催化剂,从催化剂的宏观制备方法、活性组分的浓度、载体、助剂等方面来调控Cr基催化剂的丙烷脱氢活性,并从微观结构角度深入探讨了活性位的价态、分散度、配位结构等对脱氢活性的影响,并得到以下几点较为广泛的共识:
1) 丙烷无氧脱氢的Cr基催化剂活性位是配位不饱和的低聚或隔离的Cr3+物种,该物种一部分是负载或者掺杂在载体上的Cr3+物种,另一部分是新鲜催化剂中的Cr6+物种在预还原或反应条件下转化的Cr3+物种,一般认为后者具有更高的脱氢活性;
2) 常用的载体有Al2O3、ZrO2、SiO2等惰性载体,由于Al3+与Cr3+的离子半径相近,高温反应或氧化重生过程容易使部分表面的Cr物种迁移到氧化铝体相中,是催化剂重生活性下降的原因之一,催化剂表面CrOx物种的过度聚合和积炭是催化剂失活的主要原因;ZrO2载体中的Zr4+与Cr3+的离子半径相差较大,难以大量掺杂,CrOx物种容易在表面富集;高比表面积的介孔或微孔的硅基分子筛(包括高硅铝比的微孔分子筛)为CrOx物种的高分散提供了有利条件,同时有利于丙烷与反应活性位的接触及丙烯的扩散。
3) CO2辅助的丙烷氧化脱氢有利于维持催化剂的稳定性,主要机制是消除积炭和氧化过度还原的活性位;对初试催化活性的影响取决于CO2消耗H物种促进脱氢反应平衡右移和CO2与丙烷的竞争吸附的抑制作用的博弈。
近年来,随着原位表征技术和量化计算的发展,对脱氢反应活性位的真实结构、种类的拓展及反应机理都有了新的认识和争议,本文主要总结Cr基催化剂的近期研究进展,并结合其他反应或者其他相关脱氢催化剂的研究,对Cr基催化剂长时间循环使用过程中活性位的变化、反应条件下脱氢反应活性位结构、CO2辅助脱氢过程中的决定因素及氧化脱氢机理和过渡金属氧化物助剂的作用等提出新的见解和可能性。
1 脱氢活性位及反应机理探究
丙烷无氧脱氢生成丙烯的反应机理,以SiO2表面隔离的Cr3+活性位为例,如图1所示,丙烷首先吸附并C-H键断裂生成Cr3+—C3H7和O—H中间体,随后断裂邻位C上的C—H键生成Cr3+—C3H6和Cr3+—H,丙烯脱附后2个H物种生成H2脱附,完成脱氢反应过程[5]。该机理的描述可以适用于其他类型的Cr基脱氢催化剂。
对于CrOx活性物种的CO2辅助的氧化脱氢反应机理还存在争议,该反应中同时存在脱氢反应(1)和氧化脱氢反应(2),后者涉及催化剂CrOx物种的氧化还原过程(3)和(4)。
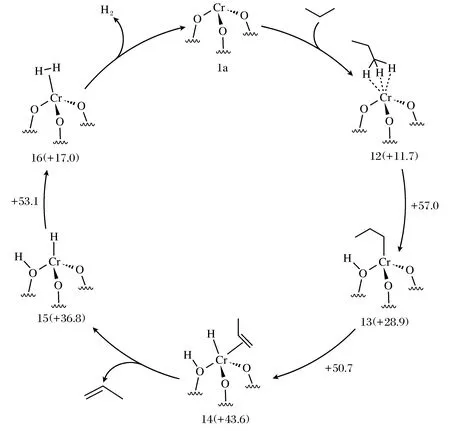
图1 SiO2表面隔离的Cr3+活性位上丙烷脱氢反应过程示意图[5]Fig.1 Schematic diagram of propane dehydrogenation for the isolated Cr3+ active site on SiO2 surface[5]
还原性气体包括丙烷及其反应衍生物(丙烯、甲烷、乙烷、乙烯、氢气等)。
Shishido等[6]通过Cr K-edge XAFS 光谱和TPR实验表明,反应初始阶段新鲜催化剂的Cr6+物种被还原为Cr3+物种;CO2能够氧化Cr3+物种为Cr6+物种,推测Cr(Ⅲ)O6与Cr(VI)O4之间的氧化还原循环催化丙烷氧化脱氢过程(图2);CO2的引入对CrOx/SiO2催化剂的脱氢反应起促进作用,对于CrOx/Al2O3催化剂则相反,归结于不同载体上CO2和丙烷的竞争吸附作用。Michorczyk等[7-9]通过原位UV-vis DRS检测反应条件下CrOx物种的价态变化,虽然CO2能够氧化部分Cr2+物种为Cr3+/Cr6+物种,但是无论丙烷反应气氛中有无CO2,Cr6+物种在反应初始阶段被迅速还原为Cr3+/Cr2+物种,Cr3+/Cr2+物种是主要的反应活性位。在该活性位上,CO2通过逆水煤气反应消耗丙烷无氧脱氢反应(1)产生H2,从而促进脱氢反应平衡右移。

图2 无CO2(a)和有CO2(b)的丙烷脱氢的反应机理[6]Fig.2 Reaction mechanism of propane dehydrogenation without CO2(a) and with CO2(b) [6]
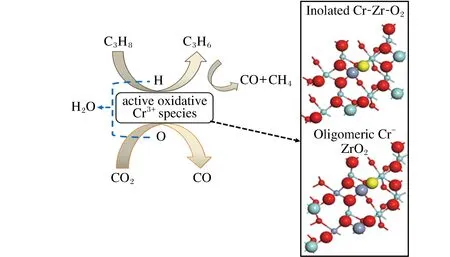
图3 xCr-ZrO2催化剂的CO2辅助丙烷氧化脱氢机理[10]Fig.3 CO2-assisted oxidative dehydrogenation mechanism of propane over xCr-ZrO2 catalyst [10]
本课题组也探究了Cr-ZrO2催化剂上的CrOx活性位上CO2辅助的丙烷氧化脱氢的过程,并提出了新的可能的反应机理。配位不饱和的低聚/隔离Cr3+物种为主要的活性位,原位拉曼光谱和反应前后催化剂的XPS结果表明诱导期后没有检测到Cr6+物种,反应过程中主要存在Cr3+物种,CO2抑制Cr3+物种还原为Cr2+物种;甲烷和CO等副产物主要是来自于中间产物丙烯与CO2在活性氧化铬物种上的反应。CO2的重要促进作用不是消除积碳,而是抑制积碳的生成,CO2消除积碳的速率很低。通过量化计算对比ZrO2掺杂Cr物种对表面晶格氧的作用发现,单位点Cr3+的掺杂显著降低Cr-O-Zr的O解离能,且低聚的Cr物种的促进作用更强,所以提出了低聚/隔离Cr3+物种中的Cr-O-Zr中的O可能为丙烷氧化脱氢的主要活性位,被命名为活性氧化铬物种,消耗O后产生的氧空位由CO2裂解出的O得以补充(图3)[10]。
2 催化剂表面Cr物种的种类及循环反应条件下的演变过程
CrOx物种在催化剂表面的价态、配位环境是多样的,受载体、负载量、制备方法、助剂等影响,普遍认为隔离、低聚、多聚(多层)、无定型的三价态的CrOx物种及晶相的Cr2O3都具有丙烷脱氢活性,但是其活性有显著的不同。以Cr2O3/Al2O3催化剂为例,新鲜催化剂主要包含以下5类Cr物种:
1) 隔离、单层/亚单层低聚的CrOx物种;
2) 可氧化还原的无定型或晶相Cr2O3颗粒表面的CrOx物种;
3) 可氧化还原的无定型或晶相Cr2O3颗粒与氧化铝键合的CrOx物种;
4) 非氧化还原的具有不同尺寸的无定型Cr2O3表面的CrOx物种;
5) 非氧化还原的具有不同尺寸的晶相Cr2O3表面的CrOx物种。
新鲜催化剂经过多次的脱氢反应和氧化重生后形成稳定催化剂,上述不同的CrOx物种经过演变并转化为以下物种:
1) 氧化铝表面隔离的CrOx物种消失;
2) 氧化铝表面弱键合的无定型Cr2O3团簇逐渐聚合长大成为直径30 nm左右的大颗粒;
3) 随着催化剂经过循环的氧化还原过程,氧化后的催化剂表面Cr6+物种减少,因为隔离的Cr6+物种的减少,只剩下无定型或晶相Cr2O3颗粒与氧化铝键合的Cr6+物种;
4) 晶相的α-Cr2O3颗粒是最稳定的物种[11]。
3 过渡金属氧化物助剂的作用
Danim等[12]在SBA-15介孔氧化硅载体上负载了0.5%Ni和10%Cr用于CO2辅助的丙烷脱氢,发现Ni的加入有利于催化活性的稳定和提高丙烯选择性,归因于Ni诱导CO2分解产生活性吸附氧物种抑制活性Cr3+物种的过度还原。通过对比Cr/Si催化剂和0.5NiCr/Si催化剂的3步H2-TPR结果发现(图4),Ni的加入显著提升了CO2氧化还原态Cr物种的能力。Li等[13]也发现Ni的修饰对氧化铝负载CrOx催化剂的丙烷无氧脱氢具有促进作用,负载的Ni物种经高温焙烧后与氧化铝形成表面分散的NiAl2O3尖晶石相,促进表面形成更多的高活性低聚Cr3+物种,且Ni的给电子作用促进Cr物种的还原性。Shao等[14]采用少量的Sn作为助剂掺杂进介孔载体氧化铝中,有效抑制生成积碳和提高脱氢活性的稳定性,归因于Sn的引入降低了氧化铝表面的强酸性、表面Cr物种的密度及表面Cr3+物种的含量。Ki等[15]探究了氧化铈修饰的CrOx-K2O-Al2O3催化剂,发现丙烯的生成速率与Ce调控的催化剂储氧能力密切相关,通过关联XPS表征和H2-TPR结果,表面Ce4+的含量与催化剂的还原性呈正比关系(图5)。

图4 Cr/Si催化剂(a)和0.5NiCr/Si催化剂(b)的3步H2-TPR[12]Fig.4 Three-step H2-TPR of Cr/Si catalyst (a) and 0.5NiCr/Si catalyst (b) [12]
上述助剂除了改变催化剂的酸碱度和表面CrOx物种的分散度,更重要的是调控了CrOx物种的氧化还原性,无论是掺杂进载体晶相中还是直接与CrOx物种相互作用,形成了新的助剂金属—O—Cr键,该新
键的形成显著影响了其催化活性。尤其是图5所示的CrOx-K2O-Al2O3催化剂体系,氧储存能力即晶格氧消耗产生氧空位和配位不饱和活性位的能力与丙烯的产率呈正比关系。Han等[16]研究表明Cr3+的掺杂ZrO2的电荷补偿作用促进生成氧空位,从而形成新的配位不饱和Zrcus脱氢活性位,该活性位上的反应机理如图6所示。
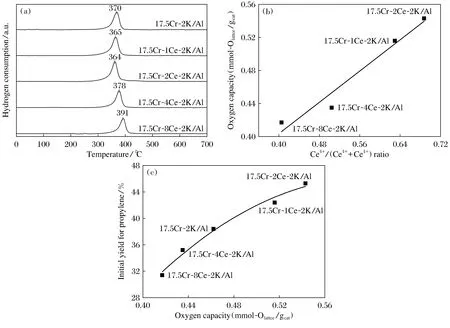
图5 (a) 17.5Cr-XCe-2K/Al(X=0、1、2、4、8)催化剂的H2-TPR图; (b) 催化剂储氧量与Ce4+/( Ce3++Ce4+)比例的关系图; (c) 催化剂储氧量与初始丙烯收率的关系图[15]

图6 含有2个氧空位的CrZrOx(101)上的能垒和反应路径示意图[16]Fig.6 Schematic diagram of the energy barrier and reaction path on CrZrOx (101) with two oxygen vacancies[16]
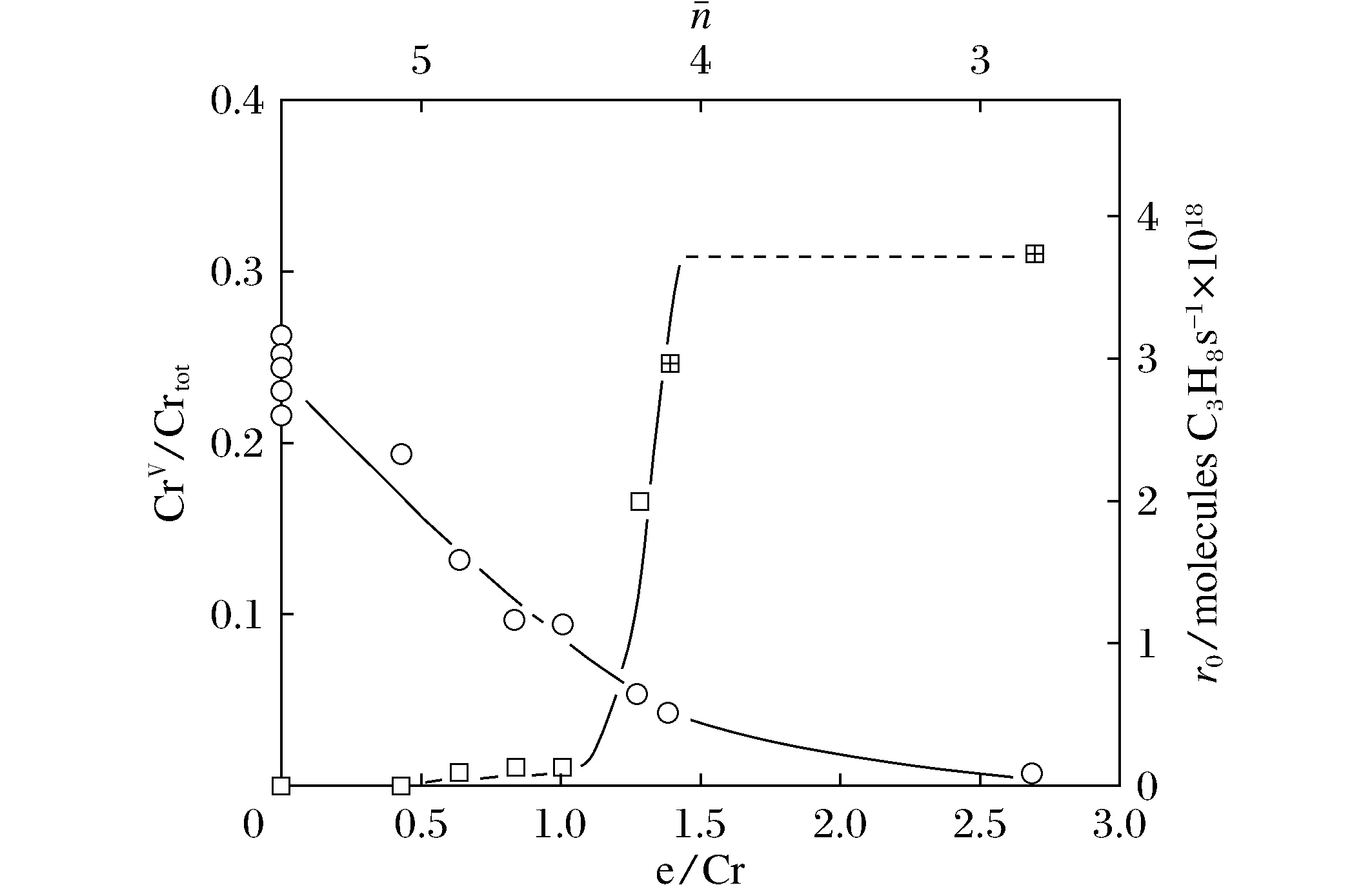
图7 CrZrOx催化剂上催化剂的还原度对丙烯加氢反应速率的影响[17]Fig.7 Effect of catalyst reduction degree on the rate of propylene hydrogenation over CrZrOx catalyst[17]

随着纳米体相氧化物丙烷脱氢催化剂的深入研究,一系列较为惰性的氧化物原本常作为催化剂载体,如ZrO2[18-19]、Al2O3[20]、TiO2[21]等,被证明还原后其表面生成的氧空位和配位不饱和的金属-氧酸碱对具有脱氢活性,该类型的催化剂表面氧空位越多其脱氢活性越高。最近也逐渐有一些文章探究金属的掺杂或表面负载,有利于特殊的晶格氧在高温还原或高温反应条件下被消耗,从而生成配位不饱和的脱氢活性位[22]。从这个角度出发,推测Cr基催化剂助剂的深层次作用,从原子层面上理解活性位与助剂或载体相互作用对脱氢活性的促进作用。
4 结论与展望
随着脱氢催化剂的表征手段,尤其是原位表征手段的发展,真实反应条件下活性位结构和反应机理被深入发掘。但是由于CrOx物种的复杂性及普遍具有脱氢活性,对关联确定的活性位与脱氢活性造成干扰。目前的研究表明,高分散的CrOx物种的配位不饱和结构的生成与稳定是催化丙烷脱氢高效和高稳定性的关键因素,推测 Cr-O-载体或助剂键中的氧缺失导致生成配位不饱和的Cr-O酸碱对是未来应该研究的重点之一。
致谢感谢沈阳市科技计划项目(Z17-5-056)和沈阳师范大学博士启动基金项目(BS201926)的支持。
猜你喜欢
大众文艺(2022年16期)2022-09-07
化学工业与工程(2022年1期)2022-03-29
中国民族美术(2021年4期)2021-07-14
能源工程(2021年1期)2021-04-13
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
今日农业(2020年20期)2020-11-26
农药科学与管理(2019年5期)2019-08-13
灾害医学与救援(电子版)(2018年1期)2018-06-05
画刊(2018年2期)2018-03-06
中国洗涤用品工业(2015年9期)2015-02-28
