
超高效合相色谱-串联质谱法定量检测食用植物油中12种长链甘油三酯
2021-05-27 03:37杨光勇郭苍亭郭金喜
分析测试学报 2021年5期
杨光勇,郭苍亭,薛 光,郭金喜
(1.新疆维吾尔自治区产品质量监督检验研究院,新疆 乌鲁木齐 830011;2.乌鲁木齐市 疾病预防控制中心,新疆 乌鲁木齐 830026)
食用植物油是人体摄取亚麻酸等生物活性物质的重要来源,亦是医药、食品加工等行业获取油脂原料的主要方式,高品质的食用植物油是确保人体营养健康和产品质量安全的重要保证。甘油三酯(Triacylglycerols,TAGs)是植物油的主要成分,不同结构的TAGs对人体的生理作用存在很大差别[1-2]。因此,准确测定食用植物油中各TAGs的含量对于评价油脂品质、保障消费者饮食营养健康等具有重要意义。
目前,油脂中TAGs的检测方法主要有高温气相色谱法[3-4]、液相色谱法[5-6]、多维色谱法[7-8]、高温气相色谱-质谱法[9-10]、液相色谱-质谱法[11-13]、高分辨质谱直接进样法[14-16]等。由于多不饱和脂肪链在高温下极易发生热裂解,因此高温气相色谱仅能用于分离饱和度较高的TAGs。液相色谱法存在污染排放高、方法开发难度大、分析时间长、分离效率较低等问题。多维色谱法能有效改善TAGs的分离效果,但操作繁杂,当使用两种不同的保留机理进行分析时,还需考虑两维溶剂的兼容性,难以实现高通量分析。高分辨质谱直接进样法分析速度快、通量高,但该法未对样品进行色谱分离,测定时各TAGs会相互干扰,影响定量结果的准确性,且所用设备较为昂贵,不易普及。超高效合相色谱(Ultra performance convergence chromatography,UPC2)是以超临界CO2为主要流动相的色谱系统,具有分离效率高、分析条件温和等优势,可有效弥补以上方法的不足,已被成功应用于TAGs的定性鉴别[17-18]、脂溶性化合物的定量分析[19-20]等方面。
食用植物油中TAGs的种类繁多,使定量检测工作变得异常困难,因此现有的大多数研究均采用面积归一化法计算各组分的相对含量。但一些研究[21-22]及本实验均发现,TAGs分子上的脂肪酰碳链长度与不饱和度对其离子化效率有很大影响,如果分析时仅考虑峰面积而忽略质谱响应差异,则会使定量结果出现很大偏差,无法反映样品中各TAGs的真实含量。针对以上情况,本文首次采用UPC2-MS/MS结合外标法对食用植物油中12种长链TAGs进行检测和精确定量,各TAGs的分离效果良好,定量结果准确,为测定食用植物油中甘油酯类化合物提供了绿色环保、准确高效的新方法。
1 实验部分
1.1 仪器与试剂
UPC2-TQS超高效合相色谱-三重四极杆质谱仪(美国Waters公司),XS205DU电子天平(瑞士Mettler Toledo公司)。
1-棕榈酸-2-亚油酸-3-油酸甘油酯(PLO)、三亚油酸甘油酯(LLL)、1,2-亚油酸-3-油酸甘油酯(LLO)、1,2-亚油酸-3-棕榈酸甘油酯(LLP)、1-棕榈酸-2-油酸-3-亚油酸甘油酯(POL)、1,2-油酸-3-棕榈酸甘油酯(OOP)、1,2-油酸-3-亚油酸甘油酯(OOL)、三油酸甘油酯(OOO)、1,2-亚麻酸-3-亚油酸甘油酯(LLnLn)、1,2-亚油酸-3-亚麻酸甘油酯(LLLn)、1,2-亚麻酸-3-油酸甘油酯(OLnLn)、1,2-亚油酸-3-硬脂酸甘油酯(LLS)标准品购自美国Nu-Chek公司、Sigma-Aldrich公司、加拿大TRC公司;正己烷、甲醇为色谱纯,购自美国Thermo Fisher公司;甲酸、氨水(25%~28%)为优级纯,购自Sigma-Aldrich公司。食用植物油样品购自市场。
1.2 实验方法
1.2.1 仪器条件色谱条件:色谱柱为两支串联的HSS C18SB柱(100 mm×2.1 mm,1.8 μm);柱温为40 ℃;流动相:A为超临界CO2,B为甲醇(含0.1%甲酸);流速为1.5 mL/min。洗脱梯度程序:0~1.0 min,0.2%B;1.0~9.0 min,0.2%~4.0%B;9.0~10.0 min,4.0%~20%B;10.0~13.0 min,20%B;13.0~14.0 min,20%~0.2%B。系统背压为12.76 MPa;进样体积为5 μL。
质谱参数:电喷雾电离源,正离子(ESI+)、多反应监测(MRM)模式;离子源温度:150 ℃;去溶剂气:流速750 L/h,温度300 ℃;毛细管电压为2.0 kV;补偿液为97%甲醇水溶液(含0.2%氨水),流速为0.3 mL/min。12种TAGs的保留时间、母离子和子离子见表1。

表1 12种TAGs的保留时间、母离子及子离子Table 1 Retention times,parent ions and daughter ions of the 12 kinds of TAGs
*quantitative ion
1.2.2 标准品与样品溶液的制备标准品溶液:准确称取12种TAGs标准品各50.0 mg,分别用正己烷制成10 g/L的单标储备液;再准确吸取各单标储备液1 mL至100 mL容量瓶中,用正己烷稀释并定容至刻度,制成各组分质量浓度均为100 mg/L的混合标准溶液。上述溶液保存于-18 ℃冰箱。
样品溶液:称取充分混匀的食用植物油0.1 g(精确至0.000 1 g),用正己烷溶解并定容至100 mL,再准确吸取该溶液1 mL,用正己烷稀释至100 mL,过0.22 μm滤膜。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 色谱柱的选择12种TAGs均为长链脂肪酸酯,各TAGs之间的结构和极性极为相似,多个化合物的母离子及部分子离子的质荷比(m/z)均相同,给选择色谱柱带来一定难度。实验考察了3种填料的UPC2色谱柱:HSS C18SB(100 mm×2.1 mm,1.8 μm)、Torus 1-AA(100 mm×3.0 mm,1.8 μm)、BEH(100 mm×3.0 mm,1.8 μm)对12种TAGs分离效果的影响。其中HSS C18SB色谱柱是经过改进的反相色谱柱,对酯类化合物具有很好的保留能力;Torus 1-AA色谱柱的填料为键合1-氨基蒽的硅胶颗粒,可提供偶极、诱导偶极、π-π等多种作用,具有较好的选择性;BEH色谱柱填装了亚乙基桥杂化颗粒,对各TAGs单体间的极性差异较为敏感。结果表明,选用BEH色谱柱时各组分的色谱峰均互相包埋或形成肩峰;Torus 1-AA色谱柱无法分离互为位置异构体的目标化合物;HSS C18SB色谱柱对各TAGs的分离效果最为明显,但经优化其他色谱条件后仍无法实现POL和PLO的基线分离。因此,实验选择串联两支HSS C18SB色谱柱(总长度为200 mm)进行测定。
2.1.2 色谱柱温度及系统背压的选择色谱柱温度和系统背压的变化会改变超临界CO2的密度,从而影响其洗脱能力。通常情况下,随着柱温升高、背压减小,流动相的密度变小,化合物的保留时间延长,但柱温升高会增大分子的能量,缩短化合物的保留时间。因此,柱温对化合物的保留行为具有双重影响。分别考察了不同柱温(20、30、40、50、60 ℃)、不同背压(12.07、12.76、13.45、14.13 MPa)对12种TAGs分离效果的影响。结果表明,随着柱温升高,各TAGs的分离趋势增加,当柱温超过40 ℃后,分离趋势反而降低;各组分的分离效果并未随系统背压的改变而出现明显变化。综合考虑分离度、系统压力极限等因素,选取柱温为40 ℃,系统背压为12.76 MPa。
2.1.3 流动相改性剂的选择为改变超临界CO2对化合物的溶解和洗脱能力,需在流动相中引入适量甲醇、乙腈等溶剂作为改性剂。常用改性剂在UPC2中的洗脱能力大小为:甲醇>异丙醇>乙腈。为获得最佳色谱峰形,实验选用洗脱强度较高的甲醇作为改性剂。同时,为消除游离脂肪酸等化合物与未键合硅醇羟基的氢键作用,降低其色谱峰拖尾对TAGs测定的干扰,在甲醇改性剂中加入0.1%甲酸,并进一步优化洗脱梯度。

图1 12种TAGs混合标准溶液(5 mg/L) 的提取离子流色谱图Fig.1 Extracted ion current chromatogram of the 12 kinds of TAGs mixed standard solution(5 mg/L) peak numbers denoted were the same as those inTable 1


图2 补偿液中氨水(A)和纯水(B)的体积分数 对2种TAGs峰高的影响Fig.2 Effects of volume fraction of ammonium hydroxide(A) and water(B) in compensation solution on the peak heights of the 2 kinds of TAGs the numbers denoted were the same as those inTable 1
在优化色谱条件下,利用UPC2-MS/MS对12种TAGs混合标准溶液(5 mg/L)进行扫描,其提取离子流色谱图见图1。
2.2 质谱条件的优化
2.2.1 采集参数的确定将各标准储备液用含0.2%氨水的甲醇溶液适当稀释后,依次注入三重四极杆质谱仪,开启仪器自动优化功能,采用自动优化所得的锥孔电压、碰撞能量和驻留时间等参数。
2.2.2 补偿液组成及流速的优化补偿液由柱塞泵输送至色谱柱后端,主要用于传输化合物至质谱仪,并促使其离子化。补偿液不参与色谱分离,但其流速和组成对化合物的色谱峰形、离子化效率等均有较大影响。为获得较好的响应强度,实验选用质子化试剂甲醇作为补偿液,并在甲醇中加入添加剂以促进TAGs离子加合物的形成。张星河等[18]的研究证实,TAGs的铵加合物具有较好的质谱响应。因此,实验以不饱和度差异最大的LLnLn和OOP为研究对象,考察了含不同体积分数氨水(0.1%、0.2%、0.3%、0.4%、0.5%)的甲醇补偿液在不同流速(0.1、0.2、0.3、0.4 mL/min)下对测定结果的影响。结果表明,当补偿液流速为0.3 mL/min,氨水体积分数为0.2%时,目标化合物的色谱峰形和响应达到最佳(见图2A)。
此外,实验发现在含0.2%氨水的甲醇补偿液中加入适量纯水可进一步提高方法的灵敏度,比较了添加不同含量(1%、2%、3%、4%、5%、6%)纯水时目标化合物的峰高(图2B),可知纯水的最佳含量为3%;当纯水含量超过5%后,系统压力波动明显,设备不稳定,可能因为水的高比热容使系统动态背压阀无法正常控温,或管路中形成双相流动相所致。
2.3 方法学验证
2.3.1 线性范围、检出限与定量下限用正己烷配制各组分质量浓度均为0.030、0.035、0.10、0.25、0.75、2.5、5.0、10、20 mg/L的系列混合标准溶液,经UPC2-MS/MS测定后,以目标物的定量离子峰面积为纵坐标(y),对应质量浓度为横坐标(x,mg/L)绘制标准曲线。结果表明,12种TAGs在相应的质量浓度范围内线性关系良好,相关系数(r2)≥0.990 2。称取适量样品,按“1.2.2”处理后进行测定,以测试溶液中各目标化合物的信噪比(S/N)分别为3和10时计算检出限(LOD)和定量下限(LOQ),12种TAGs的LOD和LOQ分别为1.0~8.0 mg/g和3.0~25 mg/g(见表2)。
2.3.2 回收率与相对标准偏差称取0.05 g(精确至0.000 1 g)亚麻籽油样品,加入适量标准储备液,使处理后的最终测试溶液中LLnLn、LLLn、OLnLn、OOP的加标浓度为500 μg/L,其他TAGs的加标浓度为50 μg/L。共制备9份加标样品进行测定,得到平均回收率(n=9)为95.7%~104%,相对标准偏差(RSD)为1.5%~4.2%(见表2)。

表2 12种TAGs的线性关系、检出限、定量下限、回收率与相对标准偏差Table 2 Linear relations,LODs,LOQs,recoveries and RSDs of the 12 kinds of TAGs
y:peak area;x:mass concentration,μg/L;the numbers denoted were the same as those in
Table 1
取混合标准溶液(500 μg/L),按“1.2.1”方法连续进样6针,得到12种TAGs峰面积的RSD(n=6)为0.48%~1.9%,保留时间的RSD(n=6)为0.12%~0.96%。将上述混合标准溶液连续测定5 d,每天进样1次,得到各TAGs峰面积的RSD(n=5)为1.1%~2.7%,保留时间的RSD(n=5)为0.90%~2.3%,表明仪器的日内和日间精密度均良好。
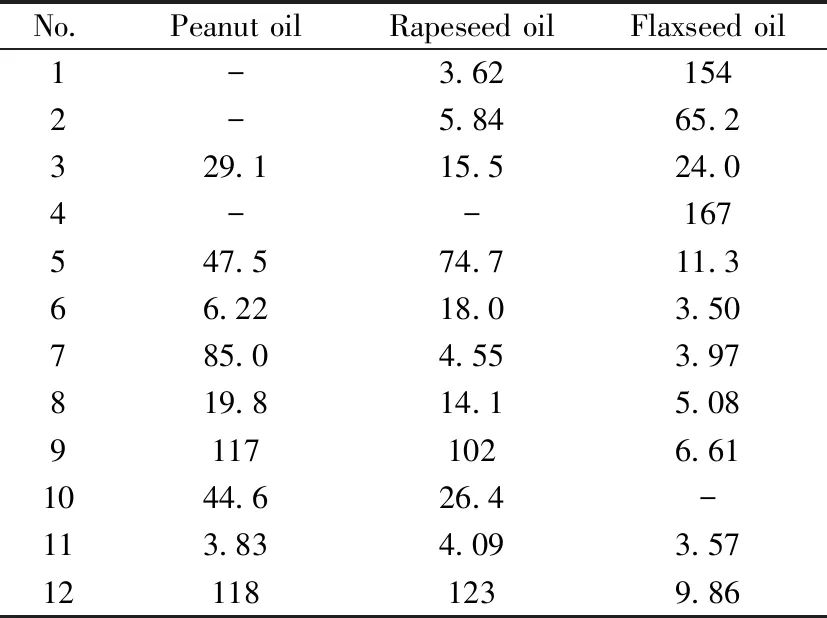
表3 样品中12种TAGs的含量(mg/g)Table 3 Contents of 12 kinds of TAGs in the samples(mg/g)
-:less than LOQ;the numbers denoted were the same as those in
Table 1
2.4 实际样品的测定
按照建立的方法对6份食用植物油样品进行分析,以外标法定量。12种TAGs在菜籽油、花生油、亚麻籽油中的含量见表3。由表3可知,各品种植物油中12种TAGs含量差异较大。其中,花生油和菜籽油中OOO的含量最高,其次为OOL,而低不饱和度的甘油三酯含量较低;亚麻籽油中则含有丰富的高不饱和度甘油三酯(如OLnLn、LLnLn等)。

3 结 论
本文采用UPC2-MS/MS建立了食用植物油中12种长链TAGs含量的检测方法,并对考察范围以外的其他TAGs进行了分析。该法快速准确,灵敏度高,专属性好,绿色环保,基质兼容性强,可有效分离当量碳数相同甚至互为位置异构体的TAGs,较传统方法极具优势,对保障植物油食用安全、评价其物理化学性质及营养价值等具有重要意义。该法也可用于食用植物油中其他酯类化合物的测定。


图3 亚麻籽油样品的提取离子流色谱图(A,m/z=869.3)及保留时间分别为4.91 min(B)、 5.03 min(C)化合物的子离子扫描图Fig.3 Extracted current chromatogram of the flaxseed oil(A,m/z=869.3),product ion scan spectra of the compound with retention time of 4.91 min(B) and 5.03 min(C)
猜你喜欢
电力安全技术(2022年3期)2022-05-30
食品安全导刊(2021年21期)2021-08-30
肉类研究(2020年9期)2020-12-14
东北大学学报(自然科学版)(2020年3期)2020-04-08
江苏农业科学(2019年7期)2019-09-17
小猕猴智力画刊(2019年5期)2019-06-21
汽车实用技术(2019年3期)2019-03-05
消费导刊(2018年15期)2018-11-24
科教导刊(2017年34期)2018-01-25
农产品市场周刊(2017年6期)2017-03-10
