
调控巨噬细胞极化的相关信号通路及其调节机制研究进展①
2021-05-26 06:13:52刘利萍张焱皓罗军敏
中国免疫学杂志 2021年6期
刘利萍 张焱皓 李 茂 秦 欢 罗军敏
(遵义医科大学免疫学教研室,贵州省免疫分子应用研究工程中心,遵义563000)
作为机体固有免疫系统的重要组成部分,巨噬细胞具有吞噬并杀伤病原微生物、免疫信息传递等功能,在炎症防御、组织发育和维持机体动态平衡等过程发挥重要作用。静息巨噬细胞在体内外不同微环境信号作用下,可发生形态、功能及表型分化,即巨噬细胞极化。根据其免疫学功能差异可将极化的巨噬细胞分为:经典激活途径的M1型巨噬细胞和替代激活途径的M2型巨噬细胞。巨噬细胞极化受信号通路、转录因子、表观遗传机制和转录后调控因子等多种分子控制,在机体微环境变化过程中发生相应的功能转换,影响疾病进程和转归,而这种功能转换与信号通路的选择性基因表达调控密切相关[1]。其中,信号通路受到广泛关注。探索巨噬细胞极化的相关信号通路调控机制有利于阐明疾病的发生发展机制,为针对性地防治疾病提供新思路。本文对调控巨噬细胞极化的相关信号通路及其调节机制的最新研究进展进行综述。
1 巨噬细胞极化
1.1 M1型巨噬细胞 在LPS和/或IFN-γ的刺激下,巨噬细胞可极化为M1型巨噬细胞,其细胞表面有MHC-Ⅱ分子、CD80、CD86等特异性标志物,以高表达IL-12和IL-23、低表达IL-10为表型特征,可产生活性氧、诱导性一氧化氮合酶(iNOS)以及生成大量TNF-α、IL-6和IL-1β等致炎因子。M1型巨噬细胞通过产生趋化因子CXCL9/MIG和CXCL10/IP-10吸引Th1淋巴细胞,并在Th1淋巴细胞介导的细胞免疫中或在活化的Toll样受体(TLR)参与下被激活,参与Th1对病原微生物的反应,在杀伤病原菌、抵抗寄生虫和肿瘤细胞及抗炎反应等生理病理过程中发挥重要作用[2]。
1.2 M2型巨噬细胞 与经典激活的M1型巨噬细胞不同,M2型巨噬细胞具有免疫调节作用,高表达抗炎细胞因子IL-10、IL-13、转化生长因子(TGF-β)等分子,促进炎症消退及病原体免疫逃逸,抑制寄生虫、促进组织修复及肿瘤发展。根据M2型巨噬细胞诱导微环境及功能特征不同,可将其细分为M2a、M2b、M2c和M2d 4个亚型。M2a型巨噬细胞由IL-4和IL-13刺激分化,高表达精氨酸酶1(Arg1)、甘露糖受体(CD206)和清道夫受体(SR),低表达iNOS,促进细胞生长、凋亡细胞清除及组织修复。M2b型巨噬细胞由免疫复合物刺激分化,高表达IL-10,低表达IL-12,促进细胞成熟,抑制炎症反应及淋巴细胞活化。M2a和M2b型巨噬细胞具有免疫调节功能,可参与Th2型细胞免疫反应。M2c型巨噬细胞由IL-10刺激分化,主要表达类几丁质酶3样分子3(Chi3L3)、穿透素3(PTX3),低表达IL-12和iNOS,抑制炎症反应,发挥免疫抑制和组织重塑作用。M2d型巨噬细胞也称为肿瘤相关巨噬细胞(TAM),抑制促炎M1型巨噬细胞,促进血管生成、伤口愈合及肿瘤细胞生长。
M1型和M2型巨噬细胞分子标记物差异显著,功能相互拮抗,但二者在特定条件下可发生表型选择性程序重排而相互转化,而巨噬细胞在维持M1/M2表型平衡过程中涉及多种信号通路调节机制。
2 调控巨噬细胞极化的相关信号通路及其调节机制
巨噬细胞极化是多因子相互作用的复杂过程,受胞内多种信号分子及其通路调控。近年关于巨噬细胞极化相关信号通路的研究不断深入,研究较为成熟的通路主要有JAK/STAT信号通路、PI3K/Akt信号通路、JNK信号通路、Notch信号通路及B7-H3/STAT3信号通路,研究表明丙酮酸激酶2(PKM2)/过氧化物酶体增殖物激活受体辅激活因子1β(TCA/PGC-1β)信号通路可能参与M2型巨噬细胞转化[3]。本文将对4条主要的信号通路及其调节机制进行综述。
2.1 JAK/STAT信号通路 JAK/STAT信号通路通过调控血细胞对各种细胞因子的反应参与细胞发育及免疫调节等重要生理过程,与肺癌、溃疡性结肠炎等多种疾病发生发展密切相关[4-5]。与细胞因子结合后,受体二聚化使其偶联的JAKs相互靠近并磷酸化各自的酪氨酸残基继而催化受体磷酸化,形成STAT停泊位点。STAT结合该位点时发生磷酸化激活形成二聚体,进入细胞核并结合特定DNA靶序列调节相应下游基因转录和表达[6]。
2.1.1 JAK/STAT信号通路介导的巨噬细胞极化 现已发现的JAKs和STATs家族成员分别包括JAK1-3、TYK2和STAT1-4、STAT5a、STAT5b及STAT6[7]。根据各细胞因子转导JAK和STAT成员的特定组合可将巨噬细胞激活的通路分为以下几种:①JAK/STAT1途径:IFN-γ通过JAK/STAT1途径促进人唾液腺导管细胞中CXCL10分泌,促使巨噬细胞发生M1型极化。而IFN-α和IFN-β通过负反馈调节抑制STAT1磷酸化,抑制M1型极化[8]。②JAK/STAT3途径:研究发现,CaMKKβ在IL-4诱导的RAW264.7细胞极化过程中通过激活JAK/STAT3信号通路促进小鼠IL-4诱导的巨噬细胞向M2表型极化[9]。③JAK/STAT6途径:IL-4与其受体结合后活化JAK,一方面直接介导STAT6磷酸化引发巨噬细胞M2型极化。同时,磷酸化的STAT6可与巨噬细胞样因子4(KLF4)和过氧化物酶体增殖物激活受体(PPAR)-γ结合促进M2型极化[10]。另一方面可通过诱导p38丝裂原活化蛋白激酶(p38MAPK)磷酸化引起巨噬细胞M2型极化[11]。巯基乙酸诱导的小鼠腹膜巨噬细胞中,IL-4诱导p38MAPK磷酸化并激活STAT6和PI3K,采用基因沉默或药理学方法抑制p38MAPK使其失活可抑制M2型极化。目前,JAK/STAT2(4和5)通路调控巨噬细胞极化研究极少,其机制研究尚未涉及,仍需进一步探索。
2.1.2 JAK/STAT信号通路的调节机制 JAK/STAT信号通路可通过自身的负反馈调节机制有效阻止细胞因子信号传导,防止生理功能紊乱,其主要负调节因子包括:细胞因子传导抑制因子(SOCS)、蛋白酪氨酸磷酸酶(PTPs)和活化STAT转录活性抑制蛋白(PIAS)。SOCS家族成员蛋白SOCS1-7可通过与JAK结合直接抑制其活性,或与STAT竞争受体结合位点抑制STAT磷酸化,泛素化降解靶标3条途径调控巨噬细胞极化[12]。SOCS1是STAT1途径的内源性抑制剂,通过其KIR和SH-2结构域启动JAK/STAT信号通路,促进巨噬细胞极化为M2型。而SOCS3作为STAT3的负调节因子,其表达上调可抑制M2型极化[13]。PIAS与活化的STAT1和STAT3特异性结合隐藏其与DNA的结合位点可抑制STAT转录,促进转录因子SUMO修饰,负向调节JAK/STAT信号通路[14]。PIPs主要通过JAK去磷酸化终止转导信号。除细胞因子调控外,蛋白化学修饰同样发挥重要作用。研究发现,IL-4通过增强JMJD3脱甲基酶表达和活性在表观遗传水平上调IRF4表达。当沉默STAT6基因时,JMJD3和IRF4表达减少[15]。最近研究发现,一种lncRNA—MacORIS与巨噬细胞极化相关[16]。MacORIS可增强IFN-γ诱导的THP-1巨噬细胞中JAK2和STAT1磷酸化,促进巨噬细胞M1型极化。
2.2 PI3K/Akt信号通路 PI3K/Akt信号通路参与细胞生长、增殖、分化及凋亡过程,其失调可引发癌症、糖尿病、心血管疾病等多种疾病[17]。某些信号因子激活I类PI3K,使第二信使磷脂酰肌醇4,5-二磷酸(PIP2)转化为磷脂酰肌醇3,4,5-三磷酸(PIP3)。PIP3与Akt的PH结构域高亲和力结合,使Akt向质膜转位并磷酸化其自身的Thr308和Ser473而激活,活化的Akt通过调控下游靶基因转录进而调节细胞功能[18]。
2.2.1 PI3K/Akt信号通路介导的巨噬细胞极化PI3K/Akt1途径介导TRAF6失活上调TLR4信号传导抑制因子IRAK-M,同时使转录因子FOXO1磷酸化失活,从而抑制TLR4的靶基因,促进巨噬细胞M2型极化。巨噬细胞Akt1缺陷(Akt1-/-)可上调miR-155表达,降低mTORC1激酶活性和Rheb水平并激活NF-κB,从而诱导M1型极化;而Akt2-/-巨噬细胞通过上调miR-146a表达促进M2型转录因子C/EBPβ和Arg1、Ym1等M2型巨噬细胞标志物表达[19-20]。miR-155不仅可通过上述途径影响巨噬细胞极化,也可抑制MyD88蛋白,促进M1型极化。但miR-155通过不同靶点改变M1/M2型极化的靶点间关联尚未明确,仍需进一步研究。mTOR作为PI3K/Akt的下游靶点,在巨噬细胞极化中也发挥一定作用。LIAN等[21]发现结肠癌细胞分泌的EGF通过结合单核细胞EGFR激活Smad-PI3K-Akt-mTOR通路,促进单核细胞极化为M2巨噬细胞。
2.2.2 PI3K/Akt信号通路的调节机制 目前,PI3K、Akt和mTOR抑制剂及PI3K/mTOR双重抑制剂的研发为PI3K/Akt通路的靶向治疗提供了可能性,可通过干预上述通路达到治疗目的,但尚未对多种分子进行深入研究。此外,还发现一些蛋白分子可调控该通路。同源性磷酸酶-张力蛋白(PTEN)是PI3K/Akt通路的上游负调节因子,其编码产物使PIP3去磷酸化生成PIP2。BAO等[22]发现miR-32通过下调PTEN激活PI3K/AKT信号通路,从而诱导M2巨噬细胞极化并调节凋亡相关蛋白表达。OHASHI等[23]发现糖原合成酶激酶3β(GSK-3β)可作为PI3K/Akt信号通路的负调控因子抑制炎症反应。由腺苷N1-氧化物激活的Akt在Ser9位点磷酸化其下游GSK-3β,活化的GSK-3β反过来抑制LPS诱导的炎症反应。LIU等[24]发现沉默信息调节因子1(Sirt1)可通过PI3K/Akt/STAT6通路改变巨噬细胞极性,抑制PI3K/Akt通路可部分阻断Sirt1的抗炎作用和STAT6易位,在Sirt1+/-小鼠中,IL-1β和Inos mRNA水平显著提高且STAT6和Arg-1表达显著降低,而激活Sirt1可上调STAT6磷酸化水平。
2.3 Notch信号通路 Notch受体在进化中高度保守,Notch信号通路功能复杂多样,参与免疫细胞发育、血管生成、胚胎发育等重要生理过程,并与肿瘤形成和部分神经系统疾病密切相关[25-26]。表达Notch受体和表达Notch配体的细胞间相互作用导致Notch信号传导级联激活,使γ-分泌酶复合物水解配体激活的Notch受体,水解的Notch细胞内结构域(NICD)通过其核定位信号易位至细胞核,结合并激活DNA结合重组信号结合蛋白Jκ影响Notch靶基因转录[27]。在没有NICD的情况下,转录因子CSL与CtBP、Hairless、SMRT和SPEN等辅阻遏物结合,抑制Notch靶基因表达[28]。
2.3.1 Notch信号通路介导的巨噬细胞极化 动脉粥样硬化是一种慢性炎症性疾病,Notch通路贯穿其各发展阶段。研究表明,DLL4/Notch1轴在促进巨噬细胞M1型极化方面起关键作用,同时阻断巨噬细胞M2型极化及其细胞因子表达[29]。NICD也可能通过直接与NF-κB蛋白结合独立于RBP-J起作用,即非经典Notch信号通路[30]。
2.3.2 Notch通路的调节机制 NICD通过翻译后修饰调节信号传导反应,现已发现多个翻译后修饰方式,包括甲基化、羟化、乙酰化、泛素化和非磷酸化。如高脂饮食诱导的肥胖通过DNA甲基化下调miR-30表达,从而在脂肪组织巨噬细胞中诱导Notch1信号传导促使其向M1型极化[31]。泛素化是最主要的调节形式,NICD的稳定性和整个信号通路的功能及其激活过程均受泛素化过程调节[32]。E3泛素连接酶Fbwx7在PEST结构域中的CDK8磷酸化位点泛素化NICD。另一种E3泛素连接酶Deltex直接结合NICD或通过蛋白Kurtz修饰NICD[33]。除表观遗传分子调节外,非编码RNA也在该通路中起调控作用。研究发现,LPS刺激后,lincRNA基因lincRNA-Tnfaip3与高迁移率族框1(Hmgb1)在巨噬细胞中组装为NF-κB/Hmgb1/lincRNA-Tnfaip3复合体,通过调节Hmgb1相关的组蛋白修饰激活NF-κB调节的炎症基因[34]。NF-κB也可通过和其他lincRNA结合调节巨噬细胞极化。研究发现,LPS刺激后,SWI/SNF复合体和lincRNA-Cox2被募集至Saa3和Ccl5基因的启动子/增强子区域,调节NF-κB的RelA和p50亚基结合到SWI/SNF复合物并促进组蛋白H3甲基化而激活其转录[35]。而XUE等[36]发现lincRNA-Cox2也可结合NF-κB的p65亚基并促进其核易位和转录,从而调节炎症小体传感器NLRP3和适配器ASC。
2.4 JNK信号通路 MAPK通路的基本组成是保守的三级激酶模式,包括MAPK激酶激酶(MKKK)、MAPK激酶(MKK)和MAPK,3种激酶依次激活。JNK信号通路作为MAPKs通路的重要分支,在调节细胞发育、炎症反应和心血管疾病发生发展等多种生理病理过程中发挥重要作用[37]。JNK的2个上游激酶MKK4和MKK7通过双磷酸化JNK的苏氨酸和酪氨酸位点特异性激活JNK,进而激活AP-1等下游底物,调节相关靶基因转录和表达[38]。
2.4.1 JNK信号通路介导的巨噬细胞极化 LPS通过TLR4/MyD88信号传导上调CCL-2表达,活化JNK导致NF-κB/AP-1转录因子活化,促进M1型极化[39-40]。PARK等[41]发 现S1P激 活ERK而 抑 制p38MAPK和JNK,并增加IL-4受体表达及促进IL-4分泌,同时通过诱导STAT6磷酸化激活SOCS1并抑制SOCS3进一步促进M2型极化。研究发现,IL-4激活的巨噬细胞中,清道夫受体1(MSR1)通过K63泛素化导致JNK活化,从而促进表型从抗炎状态转变为促炎状态,巨噬细胞向M2型转变,而在MSR1缺失时向M1型转变[42]。也有研究指出JNK通路可诱导巨噬细胞M2型极化[43]。巨噬细胞可激活M2型转录因子SMAD3从而限制M1表型形成。因此,JNK信号通路在巨噬细胞调节中表现出双重作用。
2.4.2 JNK信号通路的调节机制 JNK信号通路调节主要有2种机制,一是识别MKKK与MKK、MKK与MAPK序列,二是使三级激酶模式与支架蛋白组成复合物。MAPK通过共同对接域和谷氨酸-天冬氨酸域2个保守序列与MKK特异性结合,而一些MKK通过多功能对接位点与MKKK结合,有助于底物识别、细胞内定位、信号传导特异性和蛋白质复合物组装[44]。一些分子,如JNK抑制剂可通过竞争结合位点调节JNK通路活化。支架蛋白可促进正确的亚细胞定位,介导不同信号传递并支持多蛋白复合物形成,其本身不具有催化功能,但通过在特定结构域与其他蛋白结合改变MAPK信号通路定位位点。DIETEL等[45]发现支架蛋白PTPIP51的酪氨酸176残基在EGFR和其他激酶介导下磷酸化导致PTPIP51/14-3-3β/Raf-1复合物分解,抑制MAPK信号传导。除上述经典调节机制外,最近研究发现lincRNAEPS通过JNK/MAPK信号通路调节BCG感染的RAW264.7巨噬细胞凋亡和自噬[46]。敲低lincRNAEPS可抑制细胞凋亡并增强自噬。此外,JNK抑制剂也是JNK信号通路调节的一种重要方式。在多种疾病的动物模型中,高通量筛选所确定的特异性JNK抑制剂疗效较好,但仍需进一步临床验证。
3 信号通路的相互作用
在巨噬细胞极化过程中,虽然不同信号通路诱发的极化有所差异,但其极化途径相关,使多条信号通路间构成复杂的网络结构,通路间相互作用、相互影响,形成信号串话(图1)。
3.1 JAK/STAT信号通路与其他巨噬细胞极化相关信号通路的网络联系JAK/STAT通路与TLR通路及NF-κB通路相互交叉,研究发现多个TLR依赖于MyD88信号传导,诱导STAT1 Ser727磷酸化,TLR激活后产生肿瘤坏死因子相关受体因子6(TRAF6),TRAF6一方面直接募集并激活STAT1进而诱导巨噬细胞M1极化,一方面通过激活NF-κB通路上调IRF8蛋白表达促进巨噬细胞M1极化[47]。此外,HAMIPI等[48]发现TRAF6可多聚泛素化PI3K调节亚基p85α并促进TGF-βI型受体和p85α形成复合物,从而激活PI3K和Akt。JAK/STAT通路与JNK通路也存在相互联系。S1P可激活ERK抑制p38MAPK和JNK,而P38MAPK可抑制STAT1磷酸化。调节SIP含量及其受体表达可以调控JNK通路和JAK/STAT通路相关转录因子表达,从而影响巨噬细胞极化[41]。
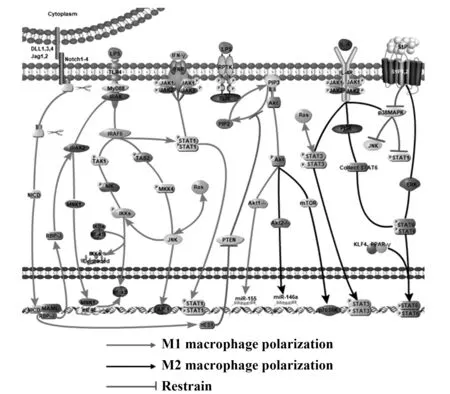
图1 巨噬细胞极化相关信号通路图Fig.1 Related signaling pathways of macrophage polarization
3.2 PI3K/AKT信号通路与其他巨噬细胞极化相关信号通路的网络联系 乳房链球菌感染中,PI3K/Akt/mTOR与TLRs/NF-κB信号通路协同促进炎症反应。TLR参与PI3K、Akt、TSC2-TSC1和Rheb同种型协同激活过程,TLRs二聚化并招募Ⅰ类PI3K及其配体Mal和MyD88导致Akt活化,进而激活下游mTOR,促进巨噬细胞M2型极化[49]。研究显示,Notch1通过HES1负调节PTEN表达诱导PI3K-Akt途径表达上调。Notch1下游的PTEN转录调节受到来自HES1的负信号和MYC的正信号双重调控。相反,PTEN突变缺失消除了这种转录调节节点,使PI3K-Akt通路与细胞外信号解偶联[50]。WEISSER等[51]发现PI3Kp110δ催化亚基可增强IL-4/STAT6转录的驱动力,而非通过诱导STAT6磷酸化直接发挥作用。
3.3 Notch信号通路与其他巨噬细胞极化相关信号通路的网络联系 NF-κB通路是TLRs和Notch通路的交集通路,TLRs激活IRAK2-MNK1-eIF4E,上调NF-κB进而促进IRF8合成。Notch通路激活后转录因子RBP-J激活MNK1,同时通过调控eIF4E与TLRs通路发生交集共同促进IRF8蛋白合成[52]。研究发现,体外感染期间Notch通路和JAK/STAT3通路存在正反馈环。由TLR4激活产生IL-6通过与其受体IL-6R结合激活转录因子STAT3,间接诱导DLL1表达,DLL1表达升高进一步激活Notch通路,产生的NICD通过上调NF-κB亚基P65促进单核细胞中IL-6分泌,IL-6阻断抗体或γ-分泌酶抑制剂可有效抑制IL-6表达及STAT3磷酸化[53]。
3.4 JNK信号通路与其他巨噬细胞极化相关信号通路的网络联系 LPS通过MyD88诱导转录因子NF-κB早期活化,并延迟诱导JAK1/STAT1活化。JNK可通过调节JAK1/STAT1通路促进巨噬细胞M1型极化[54]。T-2毒素是单端孢霉烯的主要化合物,通过激活MAPK通路抑制蛋白合成并诱导炎症和细胞凋亡。最近研究发现,T-2毒素处理的RAW264.7细胞中,JNK1和STAT3可通过K-Ras介导进行通路间串话,调节JNK通路和JAK/STAT3通路平衡可决定巨噬细胞极化[55]。
4 结语
巨噬细胞具有高度可塑性,不同环境刺激下可极化为M1型和M2型,两者极化功能几乎相互拮抗。巨噬细胞极化涉及的诱导因子、信号通路、转录因子相互交叉,形成复杂的系统,特殊条件下M1/M2可相互转化,形成动态变化过程。炎症代谢性疾病、自身免疫系统疾病及肿瘤发生发展等过程中,通过调节M1/M2型巨噬细胞极化可有效控制疾病发生发展。因此研究调控巨噬细胞极化的相关信号通路及其调节机制对各类疾病的预防、治疗和预后具有重要意义。目前对于巨噬细胞极化的信号通路和调节机制的研究不够深入和系统,由于巨噬细胞功能的多样性、信号通路转导和调控的复杂性及各信号通路的网络联系导致巨噬细胞的表型转换存在不确定性,因此需要对巨噬细胞极化的信号通路及调节机制进行进一步研究。
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07 06:21:30
现代财经-天津财经大学学报(2022年5期)2022-06-01 06:08:32
汽车维修与保养(2021年8期)2021-02-16 00:28:20
天津医科大学学报(2019年6期)2019-08-13 07:04:42
电子测试(2017年15期)2017-12-18 07:18:51
工业设计(2016年4期)2016-05-04 04:00:15
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
电源技术(2015年1期)2015-08-22 11:16:18
遗传(2014年3期)2014-02-28 20:59:01
测绘科学与工程(2014年6期)2014-02-27 07:06:21
