
北方地区单中心Citrin蛋白缺陷所致婴儿肝内胆汁淤积症23例临床特征及基因分析
2021-05-17 10:01刘文雯王美娟宁慧娟钟雪梅
临床肝胆病杂志 2021年5期
刘文雯,马 昕,王美娟,宁慧娟,钟雪梅
首都儿科研究所附属儿童医院 消化内科,北京 100020
近年来,随着分子医学及遗传代谢相关的检测技术的发展,婴儿胆汁淤积症的疾病谱正在发生转变,各种基因突变引起的遗传代谢性婴儿胆汁淤积症逐渐被认识。希特林蛋白缺陷病(citrin deficiency,CD)为一种常染色体隐性遗传疾病,最初在日本首次报道[1],在亚洲国家多见。目前认为有3种临床类型:(1)Citrin蛋白缺陷所致的新生儿肝内胆汁淤积症(neonatal intrahepatic cholestasis caused by citrin deficiency,NICCD);(2)Citrin蛋白缺陷病导致的发育不良和血脂异常(failure to thrive and dyslipidemia caused by citrin deficiency,FTTDCD)[2],是一种中间表型;(3)成人中作为复发性高氨血症伴神经精神症状的瓜氨酸血症Ⅱ型(adult-onset type citrullinemia Ⅱ, CTLN 2)。NICCD是常见的引起婴儿胆汁淤积的病因,占我国遗传代谢病第2位[3],仅次于甲基丙二酸尿症,是我国婴儿胆汁淤积性肝病重要病因。SLC25A13基因突变类型分布有明显的地域差异,北方群体的SLC25A13等位基因异质性明显高于南方群体,既往关于我国NICCD的病例及相关基因报道多来自于南方地区,本文主要分析北方地区NICCD患儿的病例特点及基因突变情况。
1 资料与方法
1.1 研究对象 收集2015年1月—2018年12月于首都儿科研究所附属儿童医院消化内科住院,并经血串联质谱、尿气相色谱和/或基因检测确诊的23例NICCD患儿的临床资料(NICCD组)。NICCD诊断标准[4]:(1)具有胆汁淤积的临床表现;(2)血串联质谱和尿气相色谱诊断;(3)基因检测示SLC25A13突变。以上3条需符合2条方可诊断,或仅有(3)同时为纯合或复合杂合致病性突变。同时收集与NICCD组同期的特发性肝内胆汁淤积症(idiopathic intrahepatic cholestasis, INC)患儿36例为INC组,即完善生化、病原、腹部影像学、血尿遗传代谢筛查、代谢性肝病相关基因panel检测等仍不能明确胆汁淤积病因的患儿。
1.2 研究方法
1.2.1 临床资料 收集患儿临床资料,包括住院时年龄、性别、尿色、大便颜色改变情况、肝脾大程度、发病时间、出生史。
1.2.2 实验室检查 收集患儿未经系统治疗前的血常规、生化、凝血、血气分析、血氨、甲胎蛋白、病原学检查、甲状腺功能检测(对于多次检测的项目取最高值)、血串联质谱(MS-MS)分析以及尿气相色谱-质谱(GC-MS)。
1.2.3 病理检查 部分病例全麻下行腹腔镜探查+胆道造影+肝活检术,术中取肝组织HE染色后光镜观察。由本院病理科完成病理切片制作,由资深病理老师阅片并出具病理报告。
1.2.4 高通量基因测序检测 住院期间留取患儿及父母外周血标本2~3 ml,置于EDTA抗凝管保存。由北京迈基诺医学检验所对胆汁淤积症相关的目标基因位点进行高通量捕获测序检测分析。
1.3 伦理学审查 本研究通过首都儿科研究所附属儿童医院医学伦理委员会批准,批号:SHERLL2020014。患儿监护人已充分知情并签署书面知情同意书。

2 结果
2.1 一般资料 23例NICCD患儿中,籍贯均为长江以北,主要来自北京市、河北省、山东省、山西省;男女比例为16∶7,均为足月儿,但出生体质量偏低,平均体质量为(2912.00±486.55)g。伴有肝外畸形者5例(21.7%),包括房间隔缺损2例,马蹄肾、肾盂肾盏扩张、隐性脊柱裂各1例。36例INC患儿中,男女比例为26∶10,其中<35周的早产儿占4例,平均体质量为(2878.00±523.49)g,肝外畸形4例,分别为房间隔缺损、小肠闭锁、十二指肠发育不良、左室假腱索各1例。临床表现比较详见表1。

表1 NICCD组与INC组临床表现比较
2.2 实验室指标比较 23例NICCD患儿中,伴有低血糖患儿10例,伴有低白蛋白血症患儿13例,伴有高血氨患儿17例,伴有高乳酸患儿15例;血脂指标中,15例LDL升高,6例TC升高,7例TG升高;凝血功能方面,17例PT延长,16例患儿活化部分凝血活酶时间(APTT)延长。与INC组对比,NICCD组GGT、TBA、APTT明显高于INC组,Alb较INC组为低,差异均有统计学意义(P值均<0.05)(表2)。

表2 NICCD组与INC组实验室检查结果对比
2.3 病理学检查 3例NICCD患儿完善肝组织活检,2例表现为肝细胞及毛细胆管淤胆,不同程度的炎性细胞浸润,肝细胞脂肪变性、气球样变,可见点灶样坏死、融合灶样坏死;汇管区轻度扩大;其中1例肝小叶结构紊乱,可见假小叶形成,散可见吞噬色素颗粒的Kupffer细胞,病变相当于G3S4。1例主要表现为汇管区小胆管数量稀小,管腔狭小。
2.4 血MS-MS和尿GC-MS检测 23例NICCD患儿中,共21例完成血MS-MS检测,主要表现为蛋氨酸、瓜
氨酸、精氨酸和游离酰基肉碱(CO)及长链酰基肉碱(C14、C16、C18∶1、C18∶2)的升高,通过血MS-MS检测诊断NICCD 10例,占47.6%(10/21)。18例完善了尿GC-MS检测,主要表现为半乳糖、半乳糖酸、半乳糖醇、4-羟基苯乳酸排泄增加。
2.5 基因检测分析 在23例患儿中复合杂合突变9例,纯合突变7例,7例为杂合突变(表3)。共检测到16种突变类型,其中IVS14-9A>G、c1640 G>A、c.762T>A、c.736delG、c.1098 T del、c.851G>A、c.550G>A为新发现突变。其中10例进行了父母验证,其中1例患儿父亲为c.615+5G>A纯合突变,余均为单个位点突变(表3)。病例6患儿为c.852_855del/IVS16ins3kb复合杂合突变,c.852_855del,导致氨基酸改变p.R284fs,为移码突变,突变来源于母亲,父亲该位点无变异;IVS16ins3kb经PCR扩增示先证者及父亲发生突变,母亲未发生突变(图1、2)。病例7患儿为c.852_855del/c.615+5G>A复合杂合突变,c.852_855del(p.R284fs)突变来源于母亲,父亲该位点无变异;c.615+5G>A(splicing)为剪切突变,受检人之父该位点纯合变异,受检人之母该位点无变异(图3)。
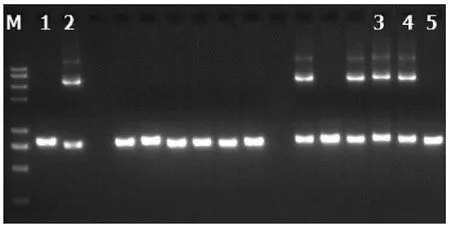
注:1,阴性对照;2,阳性对照;3,患儿;4,患儿父亲;5,患儿母亲。
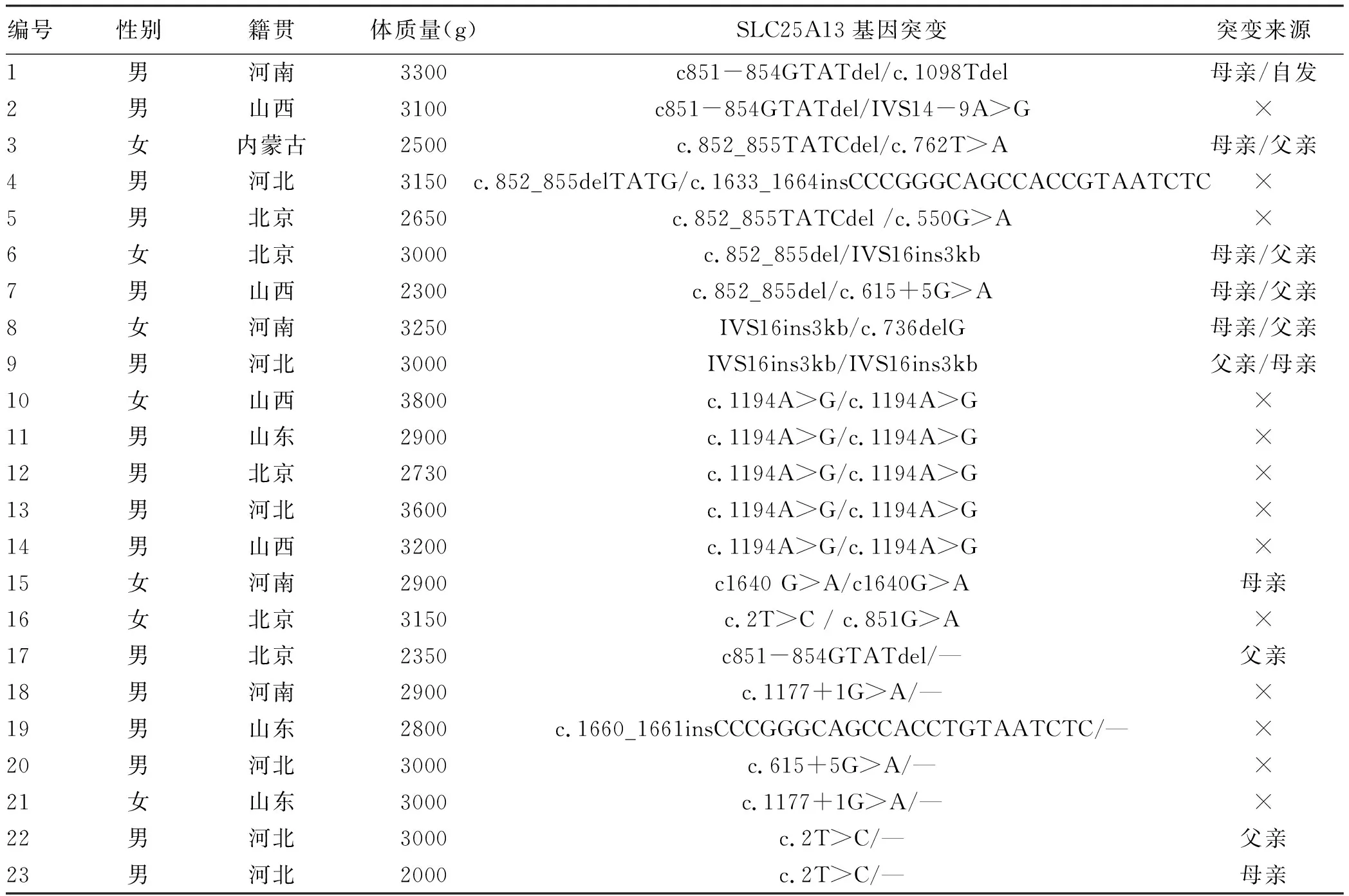
表3 23例NICCD患儿一般情况及SLC25A13基因突变检测结果

注:a,先证者杂合突变;b,父亲无变异;c,母亲杂合突变。
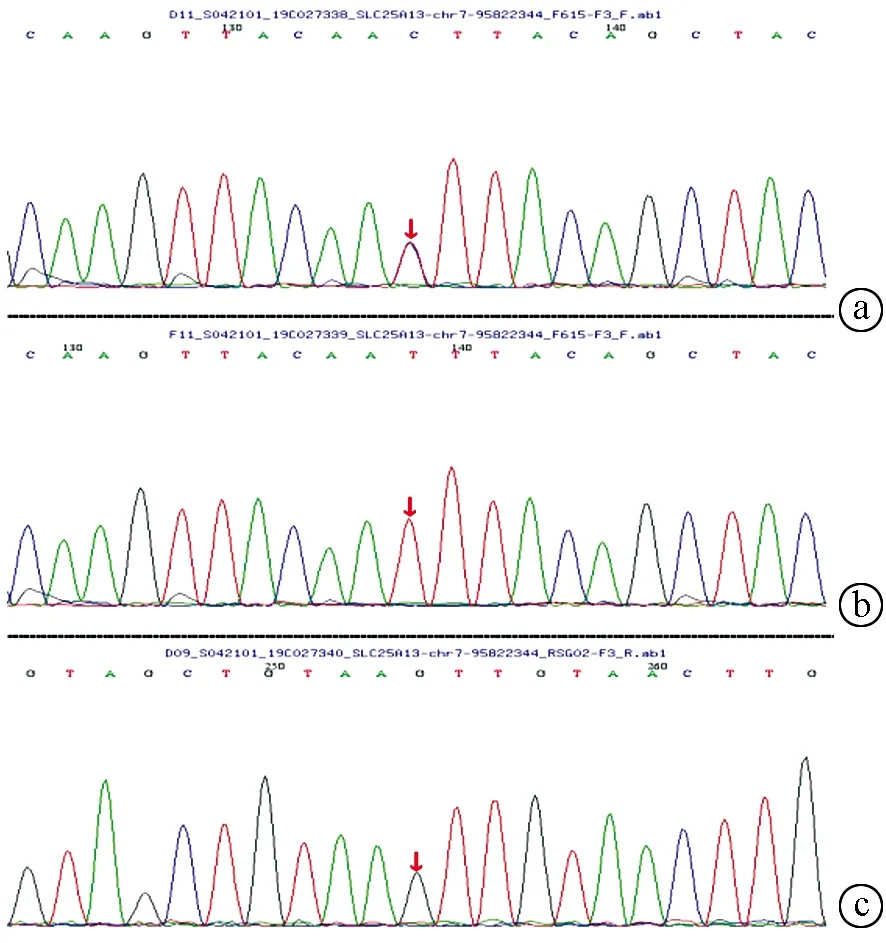
注:a,先证者杂合突变;b,父亲纯合突变;c,母亲无变异。
2.6 治疗与转归 所有患儿入院后即予保肝利胆治疗、对症补充脂溶性维生素、调节肠道菌群、更换至无乳糖并强化中链脂肪酸奶粉喂养等,部分合并感染患儿使用抗生素治疗,合并有巨细胞病毒感染者同时予以更昔洛韦抗病毒治疗。2例患儿失访,21例患儿经过治疗2~6个月后转氨酶及胆红素水平逐渐恢复正常,随访至1岁,患儿均无生长发育落后,其中2例患儿出现饮食偏好(喜食鱼类、肉类,不喜主食)。1例患儿2岁8月龄并发胆总管结石合并急性胰腺炎。
3 讨论
CD是由SLC25A13基因突变导致的一种常染色体隐性遗传疾病,致病基因SLC25A13 位于染色体7q21.3上,编码 Citrin 蛋白[1]。Citrin蛋白功能缺失时,NADH含量升高,NADH/NAD+比值升高,引起一系列代谢改变。同时,ATP产量减少,造成肝细胞能量缺乏[5]。
NICCD是我国婴儿胆汁淤积性肝病重要病因。本研究中23例NICCD中,均为婴儿期起病,其中新生儿期起病占56.5%,临床表现为不同程度的皮肤黄染、深色尿、排黄色或浅黄色大便,有13%患儿有一过性白陶土样大便,提示NICCD患儿亦可伴有严重的胆汁排泄障碍。其生化主要表现为胆红素及酶学指标的异常,同时发现胆红素及转氨酶两者的升高程度呈正线性相关,因线粒体功能障碍,苹果酸转运受阻,ATP产生减少,而胆小管膜上依赖ATP的载体蛋白转运胆红素发生障碍,造成严重胆汁淤积,从而加重肝细胞损害,造成恶性循环。转氨酶指标中AST升高幅度较ALT显著,因AST主要存在于肝细胞线粒体中,而ALT主要存在于细胞质中,因Crtrin缺陷症线粒体功能障碍,受损严重,故肝细胞膜被破坏后血AST升高较ALT更为明显。同时伴有高乳酸、血氨轻度升高、低血糖、凝血功能异常、血脂异常,这与CD所引起的糖、脂肪、氨基酸代谢异常有关。NICCD组GGT、TBA、APTT明显高于INC组,Alb较INC组低(P值均<0.05),表明上述指标可起到鉴别NICCD的作用。
血MS-MS及尿GC-MS对于CD具有重要诊断意义。本研究中CD患儿的血MS-MS检测主要表现为蛋氨酸、瓜氨酸、精氨酸不同程度的升高;除上述改变外,还有游离肉碱及长链酰基肉碱代谢异常,此为肝功能异常或胆汁淤积的非特异性改变,无明确指向性,本研究中,其阳性诊断率为47.6%(10/21),较之前学者报道[6]的32.7%稍高。部分病例血、尿代谢筛查未见异常,提示其具有一过性,曾有研究报道[7-8]指出,血氨基酸异常只出现在疾病前2个月。其尿GC-MS检测多表现为半乳糖、半乳糖醇、半乳糖酸、4-羟基苯乳酸的增高,与既往报道一致[9]。NICCD新生儿筛查敏感性较低可能与采血时间过早其代谢谱特征尚未明显呈现有关,这为本病早期诊断增加了难度。
目前已报道SLC25A13基因变异100余种,最常见的突变类型为缺失突变、错义突变、无义突变及剪接位点突变。我国报道了多种致病突变类型,包括大片段重复/缺失,Zhang等[10]通过单核苷酸多态性分析及半定量PCR技术发现了一个包含启动子及外显子1的21.7 kb缺失,完全抑制了其转录及翻译,是目前发现的最长片段缺失。我国NICCD基因突变类型以c.851_854del(占58.41%)、c.1638_1660dup23(8.85%)、c.IVS6+5G>A(8.41%)和IVS16ins3kb(7.52%)最常见,该4种突变型在我国南方占比(87.9%)大于北方(63.6%)[11-12],并且北方群体的SLC25A13等位基因异质性明显高于南方群体[13]。
本研究23例CD患儿中,包括插入突变、缺失突变、剪切位点突变及错义突变,其中高频突变c.851_854del4占3例,c.852_855del4占5例,IVS16ins3kb占3例,c.1194A>G占5例;但未见c.1638_1660dup23的突变类型,考虑与标本量少及地域性有关。其中新发现突变7种,通过多种预测软件均提示其合成的蛋白质结构为不利蛋白。多种突变类型也证实了北方群体等位基因的高异质性。本次研究中7例为单基因杂合突变,鉴于本次高通量基因测序检测方式的局限性,是否存在大片段的插入/缺失突变或尚未发现的致病突变,还需进一步研究验证。
本研究中病例5患儿合并胆总管结石伴急性胰腺炎。其6月龄诊断NICCD,基因突变类型为c.852_855TATCdel /c.550G>A的复合杂合突变,予对症保肝利胆治疗后约9月龄肝功能正常;2岁8个月出现急性腹痛症状,血生化示:血淀粉酶411 U/L,ALT 70.4 U/L, AST 49.2 U/L,胆红素正常,甘油三酯 2.56 mmol/l,腹部影像学检查提示胆总管结石征象伴胰腺肿胀,遂行ERCP下胆总管取石术以及胃镜引导下鼻-空肠管放置术,术后予鼻饲泵奶15 d,并逐渐过渡至经口喂养。随访患儿至今,目前5岁,生长发育正常,无特殊饮食偏好,肝功能正常。患儿是否发展至FTTDCD还需进一步随访监测。
病例6患儿的突变类型为第9外显子c.852_855del和第16内含子IVS16ins3kb的复合杂合突变,较为罕见,在国内仅有6例报道[14],本例患儿的蛋白质结构和功能以及其对临床预后的影响还需进一步研究,随访患儿目前1岁4个月,生长发育正常,无明显饮食偏好,检测肝功能正常。
NICCD治疗主要为保肝利胆、脂溶性维生素补充以及无乳糖并含丰富的中链脂肪酸的特殊配方奶粉治疗,饮食治疗的效果已在许多病例报告中得到证实。NICCD一般预后良好,症状可能在1岁时自行消退,但越来越多预后不良的病例相继被报道,张建玲等[15]曾报道1例以肝硬化腹腔积液起病的NICCD患儿。本研究23例中,随访的21例患儿病情均于1岁内好转,转氨酶及胆红素水平正常,无生长发育落后,其中2例患儿存在饮食偏好,关于病情是否进一步发展至Citrin蛋白缺乏症导致的FTTDCD还需进一步随访监测。存在肝外畸形的5例患者转归亦良好,目前未找到肝外畸形影响预后的直接证据。
综上所述,NICCD在婴儿胆汁淤积症中并不少见,临床医师需加强重视,对于临床上高度怀疑的病例应尽快行血尿遗传代谢性筛查,必要时完善基因检测。早期诊断,及时干预,同时对于1岁内病情缓解的患儿亦需长期进行随访,避免发展至FTTDCD甚至是CTLN2。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:刘文雯负责课题设计,资料分析,撰写论文;马昕、王美娟、宁慧娟参与收集数据,修改论文;钟雪梅负责拟定写作思路,指导撰写文章并最后定稿。
猜你喜欢
基层中医药(2022年7期)2022-11-17
基层中医药(2022年6期)2022-10-24
保健医苑(2022年5期)2022-06-10
珠江水运(2021年15期)2021-08-29
种子(2021年3期)2021-04-12
婚育与健康(2020年10期)2020-12-03
中西医结合肝病杂志(2020年2期)2020-10-27
水能经济(2017年6期)2017-10-19
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
