
缺失突变型HBx 蛋白介导PDK2 在肝细胞肝癌中的差异表达及机制
2021-05-08 06:52张晶毕利泉曹瑞雪姜贝贝张自成刘晓红
山东医药 2021年13期
张晶,毕利泉,曹瑞雪,姜贝贝,张自成,刘晓红
1 山东省立第三医院,济南250031;2 中国人民解放军联勤保障部队第960医院;3 深圳市中医院 广州中医药大学第四临床医学院
肝细胞肝癌(HCC)是我国第4 位常见的恶性肿瘤,其主要发病危险因素是乙型肝炎病毒(HBV)感染[1]。大量研究显示,乙型肝炎病毒X(HBx)基因编码的HBx 蛋白可以促进肝癌细胞的增殖、迁移和侵袭,从而加速肝癌的发展[2]。既往我们检测HCC 患者的肝癌组织及非癌性肝组织,发现HBx 蛋白羧基端的缺失突变是HBV相关HCC中的频发事件,并发现了一个HBx基因的天然缺失突变体HBx-128w,导致HBx 蛋白羧基端129-154aa 的缺失[3]。体外实验证实,缺失突变型HBx-128w较之野生型HBx具有更强的促进肝癌细胞增殖、迁移和侵袭的作用[4]。2012 年3 月—2016 年10 月,我 们 构 建 了含HBx-128w 序列的真核表达载体,以空载体pcDNA3.1 为对照组,分别转染人肝癌细胞系Huh7,利用长链非编码RNA(lncRNA)芯片筛选相关差异表达基因,并初步探讨HCC 中差异基因丙酮酸脱氢酶激酶2(PDK2)表达的调控机制。
1 材料与方法
1.1 材料 人肝癌细胞系Huh7购自中国科学院细胞库;重组表达载体pcDNA3.1-HBx-128w 购自上海锐赛生物技术有限公司;真核表达载体pcDNA3.1和脂质体LipofectamineTM2000 转染试剂盒购自美国Invitrogen 公司;鼠抗HBxAg 单克隆抗体、辣根过氧化酶标记的鼠抗兔IgG 二抗、ECL 化学发光检测试剂盒购自美国Santa Cruz 公司;兔抗PDK2 单克隆抗体购自英国Abcam 公司;十二烷基硫酸钠-聚丙烯酰胺(SDS-PAGE)凝胶配制试剂盒购自上海碧云天公司;G418 培养液、胎牛血清、DMEM 培养液、Opti-MEM 培养液、胰蛋白酶、购自美国Gibco BRL 公司;聚偏二氟乙烯膜、Western 电转移系统购自美国Bio-Rad 公司;lncRNA Array v3.0 芯片购自美国Arraystar 公司;Trizol 试剂、SuperScriptTMⅢ逆转录酶购自美国Invitrogen 公司;RNeasy Mini Kit 试剂盒购自德国Qiagen公司。
1.2 细胞转染与稳定转染细胞株的筛选 Huh7细胞培养于含10%胎牛血清的DMEM 培养液中,添加1%的青霉素和链霉素,置于37 ℃、5% CO2培养箱中,隔天换液1 次,3~4 d 传代1 次。转染前24 h 用0.25%胰蛋白酶和0.02%二乙胺四酸二钠消化对数生长期的Huh7 细胞,以每孔3×105个接种于6 孔板,随机分为pcDNA3.1 组和HBx-128w 组,每组设3个复孔,转染前1 h 更换为Opti-MEM 培养液。取1μg 待转染质粒加入100μL Opti-MEM 培养液中混匀,室温孵育10 min;取3 μL Lipofectamine 2000试剂加入100 μL Opti-MEM培养液中混匀,室温孵育5 min;将两种稀释液充分混匀,室温静置30 min,加入DMEM培养液至1 mL。弃去6 孔板培养液,每孔加入1 mL转染液,置于培养箱中培养4 h。加入含20%胎牛血清的DMEM 培养液1 mL,24 h 后更换新鲜培养液。转染48 h 后,取1/3 的细胞加入含800 μg/mL G418的培养液,筛选出抗性克隆,之后降低G418 浓度至400 μg/mL维持扩大培养抗性克隆细胞。
1.3 细胞HBx 蛋白表达检测 采用SDS-PAGE 凝胶电泳及Western blotting法。取单层生长的两组稳转Huh7细胞及未转染Huh7细胞,PBS洗2次,加入SDSPAGE上样缓冲液,冰上孵育20 min,12 000 r/min离心2 min,收集上清。98 ℃水浴变性5 min,加样至10%的SDS-PAGE 凝胶加样孔内电泳(120 V)。使用电转移系统将分离胶上的蛋白质转移至PVDF膜,10%脱脂奶粉封闭2 h,加入鼠抗HBxAg 单克隆抗体(1∶200),β-actin(1∶1 000)作为内参,4 ℃过夜。次日加入HRP 标记的二抗37 ℃孵育1 h,ECL 化学发光检测试剂盒显影成像。
1.4 细胞mRNA表达的LncRNA芯片检测 lncRNA Array v3.0 芯 片 包 括来 自Refseq、Gencode、UCSC knowngenes 等权威数据库和文献报道中的lncRNA,样本制备和芯片杂交根据制造商的标准协议进行。取稳转细胞1×106~1×107个,加入RNA 抽提试剂Trizol,裂解细胞后抽提总RNA。利用NanoDrop ND-1000 分光光度计和标准变性琼脂糖凝胶电泳检测RNA 的完整性和浓度。然后利用随机引物法,将每个样本扩增并转录成荧光cRNA。用RNeasy Mini Kit 试剂盒对标记的cRNA 进行纯化,将标记的cRNA 杂交到lncRNA 芯片上,芯片在65 ℃的滚动杂交炉Agilent Hybridization Oven 中孵育17 h。使用Agilent DNA 微阵列扫描仪扫描芯片,Agilent Feature Extraction 软件提取芯片数据,Agilent Gene Spring GX v11.5.1 软件进行原始数据的归一化和后续分析。筛选标准为Fold Change≥2.0及FDR≤0.05。
1.5 细胞PDK2 表达检测 采用RT-qPCR 方法,选取差异表达基因PDK2在稳转细胞中进行验证。利用Trizol 试剂及SuperScriptTMIII 逆转录酶进行样本总RNA的抽提及cDNA的合成,实时定量PCR反应在ViiA 7 Real-time PCR System 进行,引物设计软件Primer 5.0 设计双向引物序列如下:PDK2,F:5'- TCCAGCAATGCCTGTGAGAAA-3'和R:5'-TCGGGAAGCAGGTTGATCTC-3';GAPDH,F:5'-GGGAAACTGTGGCGTGAT-3'和R:5'-GAGTGGGTGTCGCTGTTGA-3'。根据梯度稀释DNA 标准曲线,各样本PDK2 和GAPDH 基因的浓度结果直接由机器生成,数据采用2-ΔΔCT法进行分析,重复3次。
1.6 PDK2 相关调控lncRNA 的生物信息学预测及验证 根据互作基因共表达网络寻找可能相关的lncRNA,并利用RT-qPCR 方法在稳转细胞中进行验证,引物设计软件Primer 5.0设计引物序列如下:lncRNA ENST0000511361,F:5'-TCCTGCTTAACCCTGCATTT-3'和R:5'-GGCAGTGATGTGGAGTCAGA-3'。其余步骤同前。
1.7 统计学方法 采用SPSS17.0 统计软件。计量资料符合正态分布以±s表示,两组比较采用t检验。P<0.05为差异有统计学意义。
2 结果
2.1 稳转细胞HBx 蛋白表达 HBx-128w 转染组Huh7 细胞检测到HBx 阳性条带,而pcDNA3.1 空载体组和未转染组Huh7细胞无此条带,说明HBx蛋白在HBx-128w转染组Huh7细胞内高表达。见图1。
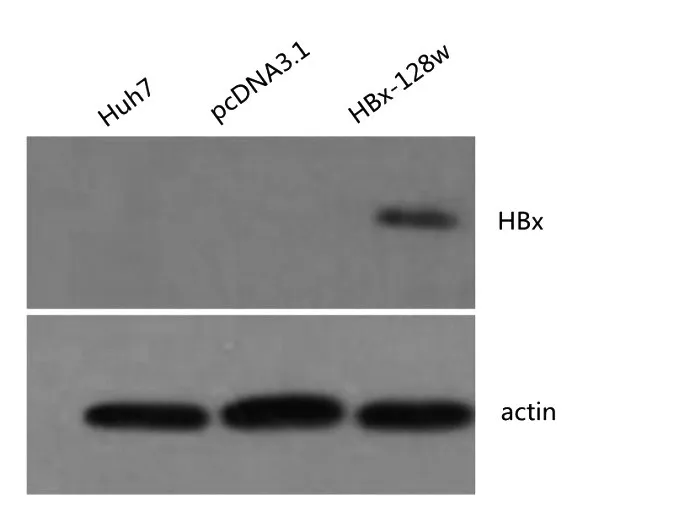
图1 两组细胞HBx蛋白SDS-PAGE凝胶电泳图
2.2 两组细胞mRNA 的差异表达 HBx-128w 组及空载体pcDNA3.1 组稳转细胞中共检测出mRNA 24 976个,lncRNA 36 629个。与pcDNA3.1组相比,HBx-128w 组差异表达的mRNA 数量为2 937 个,其中上调1 123 个(Fold Change:2.01~15.81),下调1 814 个(Fold Change:2.00~92.17)。 与pcDNA3.1 组相比,HBx-128w 组差异表达的lncRNA 数量为8 057个,其中上调2 986个(Fold Change:2.00~2578.19),下 调5 071 个(Fold Change:2.00~99.98)。在这些差异表达的mRNA中,PDK2显示为上调表达,Fold Change为3.219。
2.3 两组细胞PDK2 差异表达 RT-qPCR 结果显示,HBx-128w 组和pcDNA3.1 组PDK2 的相对浓度分别为4.823±0.497 和1.343±0.061;与pcDNA3.1组相比,HBx-128w 组PDK2 的浓度升高(P<0.05),两组间差异倍数为3.591。
2.4 PDK2 相关调控lncRNA 的差异表达 RT-qPCR 结果显示,HBx-128w 组和pcDNA3.1 组lncRNA ENST0000511361 的相对浓度分别为4.790±0.364和1.280±0.066,与pcDNA3.1 组相比,HBx-128w 组lncRNA ENST0000511361的浓度升高(P<0.05),两组间差异倍数为3.742,与PDK2差异表达的趋势一致。
3 讨论
研究表明,HBx 蛋白的氨基端(1-50aa)是负功能调控区,可以抑制HBx 蛋白的反式激活功能,而羧基端(51-154aa)为反式激活区,该区域的105-148aa是HBx 蛋白发挥反式激活功能的结构基础[5]。肝癌组织中普遍存在着异质的HBx 羧基端缺失突变体,表达截短的HBx 蛋白,导致其反式激活能力下降,同时丧失了野生型HBx 抗增殖和促细胞凋亡以及抑制细胞转化的作用[6]。HBx 蛋白任何区域的缺失都可能改变其与细胞增殖、转化、反激活或转录调控相关的生物学功能。之前我们构建了不同长度的HBx 羧基端缺失突变体,结果显示它们对肝癌细胞的生物学行为有着不同的影响[7]。对人HCC组织中HBx 羧基端缺失突变体的研究显示,与野生型HBx 相比,缺失突变体通过复杂的分子调控网络促进了肝细胞的恶性转化[8],但是其确切的分子机制仍需深入研究和充分阐明。
相关研究表明,即使在氧含量正常的情况下,肿瘤细胞也以糖酵解途径作为主要的能量来源[9]。丙酮酸脱氢酶激酶(PDKs)作为有氧糖酵解途径的重要调节酶,在某些病理条件下被激活,可能通过影响有氧糖酵解改变细胞的代谢,参与细胞的恶性转化。PDKs 有PDK1、PDK2、PDK3、PDK4 四种亚型,其中PDK2 几乎在所有组织中表达,尤其是肝脏和肾脏[10]。近年来,PDK2 与肿瘤的关系受到关注。研究显示,PDK2 在多种肿瘤中异常过表达,与肿瘤的恶性表型密切相关[11]。一项研究发现,PDK2 在胆管癌组织中强阳性表达,在癌旁组织中呈弱阳性表达,两者有显著差异[12]。本研究的lncRNA芯片结果显示,PDK2 在缺失突变型HBx 转染的Huh7 细胞中上调表达;之后的RT-qPCR 试验结果显示,与空载体pcDNA3.1 组相比,HBx-128w 组稳转细胞中PDK2 的表达显著上调,这与芯片结果一致,提示HCC 组织中,缺失突变型HBx 参与调控了PDK2 在转录水平的表达,可能通过改变肝癌细胞的代谢,加速了HCC的演进。
PDK2 调控肿瘤发展的机制涉及多个方面。有研究发现,PDK2 在HCC 中高表达,与miR-214 呈负相关,下调PDK2 可以显著抑制HCC 细胞的增殖和迁移[13]。还有人发现,抑制PDK2 活性可以抑制磷脂酰肌醇3 激酶-蛋白激酶B 通路的激活,从而抑制肿瘤细胞的生长[14]。现有对PDKs 抑制剂的研究发现,PDK2 的过表达是癌细胞获得性耐药的重要机制,下调PDK2 能够提高癌细胞对化疗药物的敏感性[15]。lncRNA 是一种长度超过200 个核苷酸、编码蛋白能力较低或不具有编码蛋白能力的RNA 转录本[16],具有复杂的二级空间结构,可以为蛋白质提供多个结合位点,形成复杂精细的基因表达调控网络。现有研究表明,lncRNA 与人类肿瘤密切相关,lncRNA 的异常表达在包括HCC 在内的许多肿瘤中都有发生[17]。目前已发现多种lncRNA 在HBV 相关的HCC中异常表达,参与调控HCC的发展[18]。大部分研究也表明lncRNA受到HBx的调控;通过加速肿瘤细胞的增殖、侵袭和转移,或通过调节多种蛋白编码基因、miRNA 和信号通路的表达来减少细胞凋亡,从 而 促 进HCC 的 进 展[19-20]。本 研 究 通 过 对mRNA 和lncRNA 表达谱的交集分析和生物信息学预测,发现lncRNA ENST0000511361 可能参与HCC发展过程中PDK2 表达的调控,RT-qPCR 结果显示HBx-128w 组稳转细胞中lncRNA ENST0000511361表达量较pcDNA3.1 组明显升高,这与PDK2 的RTqPCR 结果一致。未来我们将通过体外实验对该lncRNA的功能进行深入研究。
综上所述,我们利用lncRNA芯片筛选了缺失突变型HBx 转染的肝癌细胞中差异表达的基因,发现了PDK2 在Huh7 细胞中上调表达,并通过RT-qPCR验证了芯片结果。提示HCC 组织中,HBx 蛋白可以通过羧基末端的缺失突变,改变其生物学功能,调控PDK2 的异常表达,促进HCC 的发展。这一过程涉及到许多因素,其中lncRNA ENST0000511361 可能是一个重要的调控靶点。
猜你喜欢
湖南畜牧兽医(2021年6期)2022-01-24
中学生物学(2021年8期)2021-11-02
食品安全导刊(2021年21期)2021-08-30
昆明医科大学学报(2021年4期)2021-07-23
猪业科学(2021年5期)2021-06-02
世界科学技术-中医药现代化(2021年10期)2021-03-02
中西医结合肝病杂志(2020年2期)2020-10-27
山西农业大学学报(自然科学版)(2020年1期)2020-03-04
质量安全与检验检测(2020年6期)2020-02-01
中国果业信息(2019年1期)2019-01-05
