
射频磁控溅射制备Mn 掺杂ZnO 薄膜的结构和铁磁性能研究
2021-04-16 05:43阳生红蒋志洁张曰理
电子元件与材料 2021年3期
阳生红,蒋志洁,张曰理
(1.中山大学物理学院,广东广州 510275;2.中山大学材料科学与工程学院,广东广州 510275)
稀磁半导体(DMS)是通过在半导体中掺杂磁性离子而获得铁磁性(FM)的一种新型材料。其中磁性离子掺杂ZnO 是最有前途和研究最广泛的DMS 之一[1-4]。自从Ditel 等从理论上预言Mn 掺杂ZnO 可能表现出室温铁磁性(RTFM)以来[4],许多研究小组相继从理论和实验上证实了过渡元素(TM)掺杂ZnO 是制备DMS 的有效方法[1-3,5-6]。然而,关于TM 掺杂ZnO 稀磁半导体中掺杂磁性离子的作用仍然存在很多争议,甚至出现了相互矛盾的结论。DMS 的FM 起源也具有争议性[1-3],有人认为是由金属团簇、缺陷引起的。这是目前研制RTFM 的ZnO 基DMS 的主要障碍。众所周知,ZnO 中的本证缺陷有氧空位(VO)、间隙锌原子、锌空位以及间隙氧原子。Mn 因其3d 壳层具有最多的未成对电子(3d5),从实验和理论的角度研究Mn 掺杂ZnO 的FM 引起了研究者的广泛兴趣[1-6]。Mn 掺杂ZnO 的FM 主要认为来源于晶格缺陷和二价Mn 离子替代Zn 离子。也有研究报道Mn 掺杂ZnO 材料中的FM 来自于Zn 和O 晶格位上的缺陷[6-8]。一定量的Mn 掺杂能使居里温度提高到室温[4]。此外,氧化锌中Vo浓度的变化与FM 的强弱变化是一致的,说明FM 是基于Vo诱导机制[9]。大部分实验研究认为Mn 掺杂ZnO 材料中的FM 属于样品的本征磁性。当然,Mn 掺杂ZnO 的FM 对制备条件和制备方法十分敏感。这是因为不同的制备方法会导致材料中产生的缺陷不同所致[5,8]。实现室温铁磁性及探索其磁性来源是当今DMS 的重要研究方向。近几年,Mn 掺杂ZnO 基DMS 的研究取得了巨大的进步。但不同的制备方法、温度、气氛以及材料的结构形态都会引起磁性能的变化,开展不同制备条件和制备方法研究,对理解Mn 掺杂ZnO 的FM 来源是非常有意义的。射频磁控溅射法可以更好地控制沉积参数(如:沉积速率、衬底温度、工作气压等),并具有比脉冲激光沉积法(PLD)和分子束外延法(MBE)相对简单和低成本等优点,是制备Mn 掺杂ZnO 薄膜普遍采用的方法之一。关于溅射参数(如工作气压、衬底温度、射频(RF)源或直流(DC)源、衬底旋转速度和衬底材料等)对ZnO 或掺杂ZnO 薄膜的结构和性能的影响已有大量文献报道[10-12]。本文采用射频磁控溅射法制备了不同浓度Mn 掺杂ZnO 薄膜,研究了Mn 掺杂浓度对薄膜结构和磁性能的影响。并对Mn 掺杂ZnO 薄膜中FM 的来源进行了讨论。
1 实验
实验使用的靶材为直径两英寸的Mn 掺杂ZnO(Zn1-xMnxO,其中x=0,0.03,0.06)陶瓷靶,购自合肥科晶材料技术有限公司。采用射频磁控溅射法在Si(100)衬底上制备了Zn1-xMnxO(x=0,0.03,0.06)薄膜。在镀膜之前,Si(100)衬底依次经无水乙醇,60℃丙酮溶液超声清洗10 min,无水乙醇浸泡20 min,然后去离子水反复冲洗后在真空干燥箱里烘干。将清洗之后的衬底置于溅射室中,系统真空抽至6.5×10-6Pa,然后充入高纯(99.999%)氩气和氧气,保持Ar 流量为26 mL/min、O2流量为4 mL/min(即氩氧体积比为26 :4)的条件不变,使溅射室工作气压为2.5 Pa。调整衬底与靶间距离为7 cm,等衬底温度达到700 ℃时,首先进行预溅射15 min,以清除靶材表面的污染物。然后将溅射功率调至100 W,保持工作气压、靶材与衬底的距离、氩氧比、溅射时间(120 min)以及衬底温度(700 ℃)等参数不变,分别对不同Mn 掺杂浓度的ZnO 陶瓷靶进行溅射,制备出Zn1-xMnxO(x=0,0.03,0.06)薄膜样品。用台阶仪测得薄膜厚度约为230 nm。
用日本理学(RIGAKU)X 射线粉末衍射仪(DMAX 2200 VPC03030502)和英国雷尼绍公司生产的激光显微拉曼光谱仪(Renishaw inVia)研究薄膜的相结构、结晶取向和结晶度。用布鲁克(Bruker)公司生产的扫描探针显微镜(Dimension Fastscan)研究样品的表面形貌。采用美国Quantum Design 磁学测试系统(SQUID 磁强计MPMS3)对样品进行磁性测量。
2 结果与讨论
2.1 Mn 掺杂浓度对Zn1-xMnxO 薄膜结构的影响
图1 为不同浓度Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜样品的XRD 图谱。从图1 可以看出,未掺杂和Mn 掺杂Zn1-xMnxO 薄膜样品均呈现显著的(002)择优取向,在整个衍射角范围内未发现其他衍射峰出现。这表明未掺杂和Mn 掺杂Zn1-xMnxO 薄膜样品均具有很好的六角纤锌矿结构,属P63mc 空间群。Mn掺杂后未出现金属Mn、氧化物和其他第二相。Mn 掺杂ZnO 仍保持ZnO 的六角纤锌矿结构,说明Mn 原子替代了Zn 原子晶格位置,属替位式掺杂。随着Mn 掺杂量的增大,(002)衍射峰的强度减弱,峰位略向小衍射角方向移动(如图1(b)所示)。这表明Mn 掺杂不仅导致薄膜的结晶质量下降,而且使(002)晶面的间距增大。导致薄膜结晶质量下降的原因可能是因为Mn 的掺入导致一些缺陷(如:氧空位、锌间隙原子、边界位错及堆垛层错等)所致,而Mn 的掺入使(002)晶面间距增大是由于Mn 离子半径(80 pm)大于Zn 离子半径(74 pm)所致。Mn 掺杂量x为0,0.03 和0.06的薄膜样品,其(002)衍射峰的半峰宽(FWHM)分别为0.523°,0.541°和0.763°,随着Mn 掺杂量的增加而增加。利用Debye Scherrer 公式D=0.89λ/(βcosθ)(这里β为(002)衍射峰的FWHM,θ是布拉格衍射角,λ=0.154 nm,为X 射线的波长)对不同浓度Mn 掺杂Zn1-xMnxO 薄膜的晶粒大小进行了估算。对Mn 掺杂量x为0,0.03 和0.06 的Zn1-xMnxO 薄膜,计算得到的晶粒尺寸分别为14.2,12.4 和9.5 nm。可见晶粒尺寸随Mn 掺杂量的增加而减小。
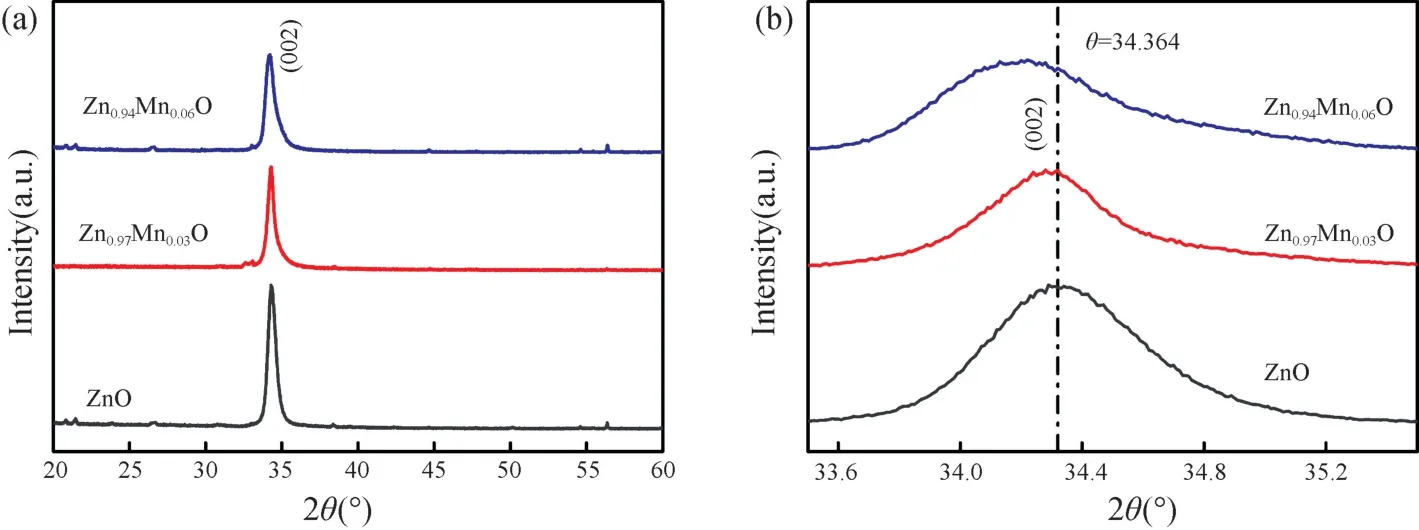
图1 (a) 不同浓度Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜样品的XRD 图谱;(b)(002) 衍射峰的放大图像Fig.1 (a) XRD patterns of Zn1-xMnxO(x=0,0.03,0.06)thin films;(b) The enlarged (002) peaks of Zn1-xMnxO(x=0,0.03,0.06)thin films
图2 为不同浓度Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜样品在波长为514 nm 的激发光下测量得到的拉曼(Raman)光谱。从图2 中可以看到Raman 特征散射峰主要出现在200~600 cm-1区域内。所有薄膜样品在437.6 cm-1位置显现出一个Raman 散射峰,该峰为E2(High)振动模式,是六角纤锌矿结构ZnO 的特征Raman 散射峰,该Raman 散射峰表明Zn1-xMnxO(x=0,0.03,0.06) 薄膜均形成良好的六角纤锌矿结构[12],这与XRD 结果是相吻合的。在488.8 cm-1和590.2 cm-1位置呈现出的两个Raman 散射峰与Zn1-xMnxO(x=0,0.03,0.06)薄膜中的氧空位、Zn间隙原子以及表面或界面光子有关[13],是缺陷引起的本征振动。峰的强弱表明薄膜样品中的氧空位、Zn 间隙原子等缺陷的多少。未发现Mn 氧化物和其他杂相相关的Raman 散射峰,说明Mn 掺杂已经进入到ZnO晶格中并替代了Zn 离子。这一点也是与XRD 结果相一致的。基于 Raman 光谱可以认为:Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜样品均具有良好的ZnO 六角纤锌矿结构,薄膜中存在一定量的晶格缺陷。
图3 为不同浓度Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜表面的二维和三维AFM 形貌图。所有样品的晶界都很明显,表面比较平整且粗糙度比较低,晶粒沿一个方向生长。从二维AFM 形貌图中可以看到,薄膜表面为分布均匀的多孔结构,孔径大小随Mn 掺杂量x的变化而变化。当x为0,0.03 和0.06 时,相应的表面均方根粗糙度分别为14.5,7.98 和3.29 nm。三维AFM 形貌图为中空的锥柱状结构,高低起伏均匀。AFM 分析表明,随着Mn 掺杂量的增加,晶粒尺寸逐渐变小,说明Mn 掺杂已进入ZnO 晶格并且通过替位方式改变了ZnO 的结构。
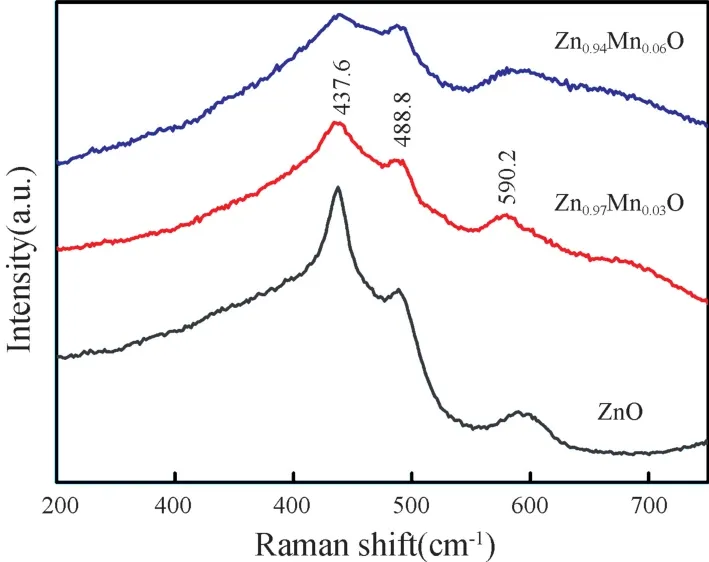
图2 不同浓度Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜的拉曼光谱Fig.2 Raman spectra of Zn1-xMnxO(x=0,0.03,0.06)thin films
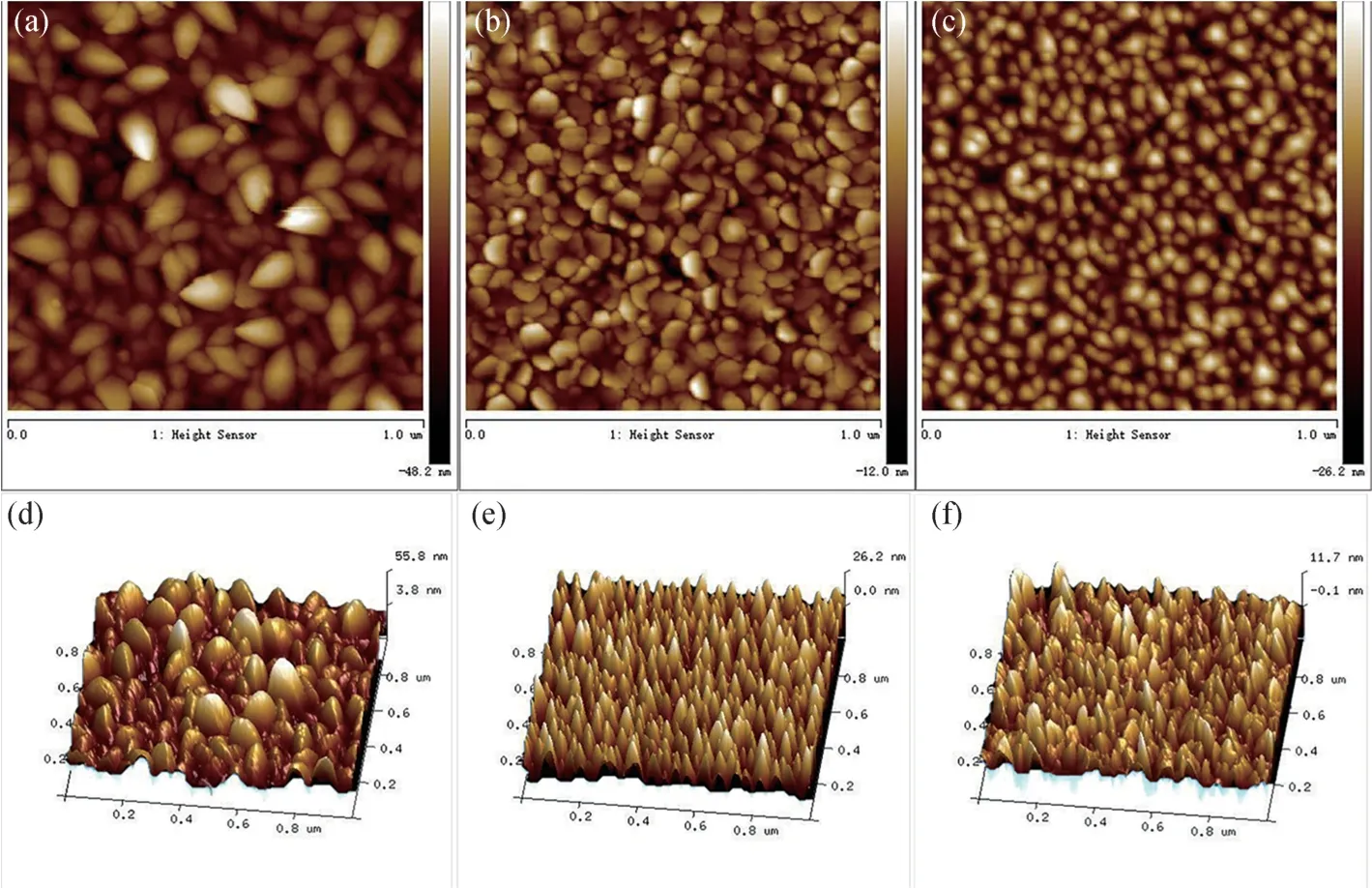
图3 不同浓度Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜表面的二维和三维AFM 形貌图。(a)、(d)x=0;(b)、(e)x=0.03;(c)、(f)x=0.06Fig.3 Two and three dimensional AFM images of Zn1-xMnxO(x=0,0.03,0.06)thin films.(a) and (d)x=0;(b) and (e) x=0.03;(c) and (f)x=0.06
2.2 Mn 掺杂浓度对Zn1-xMnxO 薄膜磁性能的影响
图4 是不同浓度Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜样品的室温磁滞(M-H)回线。由图4 看出,未掺杂ZnO(x=0)薄膜样品在室温下只显示出顺磁性,当Mn 掺杂ZnO 薄膜样品在室温下显示出明显的磁滞回线,具有室温铁磁性。对Mn 掺杂量x为0.03 和0.06 时,矫顽力(Hc)和1.59×106A/m 磁场下的磁化强度(M)分别为2.16×104A/m,0.12 A·m2/kg 和2.99×104A/m,0.18 A·m2/kg。Mn 掺杂Zn1-xMnxO薄膜样品的室温铁磁性随着Mn 掺杂量的增加而加强。这是由于随着Mn 掺杂量的增加,替代Zn 的Mn 离子增多,导致铁磁交换作用增加的结果。
对于掺杂ZnO 薄膜样品的铁磁性起源问题仍存在争议,一方面人们认为样品的铁磁性来源于金属团簇或第二相,另一方面认为是样品掺杂的本征性质。本工作中制备的Mn 掺杂Zn1-xMnxO 薄膜样品不难排除Mn 团簇、Mn 的氧化物和可能的第二相对薄膜样品铁磁性的贡献。从XRD 和Raman 图谱可以看出,所有样品均保持良好的ZnO 六角纤锌矿结构,未出现其他杂相。可排除薄膜样品存在Mn 团簇、Mn 的氧化物和可能的第二相的情况。因此,本工作中制备的Mn 掺杂Zn1-xMnxO 薄膜样品的铁磁性为材料的本征性质,即Mn 替代Zn 的铁磁交换和反铁磁交换的竞争。很多报道认为氧空位对Mn 掺杂ZnO 和Co 掺杂ZnO 薄膜的铁磁性起着重要的作用[14-15]。考虑到所采用的制备方法和结构分析的结果,本工作中制备的Mn 掺杂Zn1-xMnxO 薄膜样品虽然具有良好的ZnO 六角纤锌矿结构,但薄膜内也同时存在一定数量的晶格缺陷(如:氧空位、Zn 间隙原子等)。根据束缚磁极化子(Bound Magnetic Polaron)模型[16-17],当氧空位浓度达到一定值时,邻近的磁极化子相互交叠,从而导致邻近的Mn离子之间产生铁磁交换作用。这是Mn 掺杂Zn1-xMnxO薄膜样品中观测到室温铁磁性的主要原因。随着Mn掺杂量的增加,氧空位或缺陷均有所增加,从而提供了更多的铁磁耦合中心,故样品表现出更强的铁磁性。
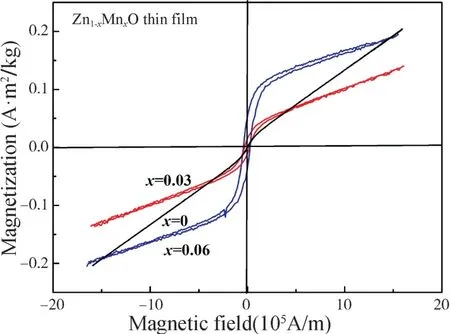
图4 不同Mn 掺杂Zn1-xMnxO(x=0,0.03,0.06)薄膜样品的室温磁滞回线Fig.4 Room temperature M-H curves of Zn1-xMnxOthin films with x=0,0.03,0.06
3 结论
利用射频磁控溅射法制备了不同浓度Mn 掺杂Zn1-xMnxO 薄膜,并采用XRD、Raman、AFM 和SQUID 对其结构、表面形貌和铁磁性进行了研究。XRD 和Raman 研究没有发现第二相,所有薄膜样品均保持良好的ZnO 六角纤锌矿结构,且沿c轴择优取向生长。AFM 测量表明样品中的颗粒分布均匀,没有出现团聚物。SQUID 测量结果表明,未掺杂ZnO 薄膜在室温下只显示出顺磁性,而Mn 掺杂ZnO 薄膜在室温下显示出明显的铁磁性,且随着Mn 掺杂量的增大,样品的矫顽力(Hc)和磁化强度(M)均增大。基于XRD、Raman 和AFM 研究结果,可以认为本工作中发现的Mn 掺杂ZnO 薄膜的室温铁磁性是其本征性质,其室温铁磁性的来源可通过束缚磁极化子模型解释。
猜你喜欢
华北地震科学(2022年3期)2022-07-22
矿产保护与利用(2022年1期)2022-05-05
银潮(2021年12期)2022-01-18
矿产勘查(2020年5期)2020-12-25
陶瓷学报(2019年5期)2019-01-12
中国有色冶金(2018年4期)2018-01-31
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
浙江电力(2014年6期)2014-01-27
铜业工程(2010年1期)2010-09-14
