
先天性无痛无汗症家系致病基因的研究*
2021-04-08 01:40刘智鸿李胜高德海刘世祺
中国医学工程 2021年3期
刘智鸿,李胜,高德海,刘世祺
(1.山东省药物研究院,山东 济南 250062;2.济宁医学院附属医院 胃肠外科,山东 济宁 272029)
遗传性感觉和自主神经病Ⅳ型[又称先天性无痛无汗症(congenital insensitivity to pain with anhidrosis,CIPA)]是一种非常罕见的常染色体隐性遗传病[1],发病率约为1/25 亿。1963 年SWANSON[2]将其正式命名为CIPA。DYCK[3]于1983 年将伴有痛觉缺乏的各种疾病命名为遗传性感觉和自主神经病(hereditary sensory and autonomic neuropathy,HSAN),并根据遗传方式和受累神经细胞或轴突的数目进行分型(共分为5 型),CIPA被定义为遗传性感觉和自主神经障碍(HSAN)Ⅳ型。
CIPA 患者常见临床表现全身无汗、对疼痛刺激无反应,出现反复发作不明原因的发热、感染[4],CIPA 患者还会出现不同程度的骨关节病变、关节肿胀[5]、关节囊松弛等病变、Charcot 关节病[6-7]及脊柱病等。CIPA 患者还存在一定程度的智力发展缺陷[1]。多数观点普遍认为,CIPA 诊断需综合多项资料进行分析:①明确无痛觉、无汗病史及症状;②痛觉、温度觉试验及碘淀粉法发汗定性试验是该病症的诊断依据;③皮肤活检病理有助于确诊,皮肤组织结构及汗腺形态正常或萎缩,周围神经无髓鞘及细小有髓鞘纤维丢失等[1];④检测到NTRK1 基因突变则高度支持CIPA 诊断。符合以上条件,结合患者的具体表现,可以确诊为CIPA。在国外,CIPA 患者中已有神经营养因子酪氨酸激酶受体1 型(NTRK1)为其主要致病基因报道[8-13]。INDO 等[14]于1996 年已将NTRK1 基因突变确定为CIPA 的致病原因。
本研究收集的一个CIPA 家系的基础上,采集患者皮肤及皮下组织,进行HE 染色及免疫组织化学SP 法,观察皮肤是否神经末梢缺如,检测S-100 蛋白表达,明确该CIPA 患者诊断,进一步采集该CIPA 家系成员外周血,分离白细胞、提取DNA;通过外显子测序方法,检测该家系NTRK1基因和FAM134B 基因的突变情况,分析其结果与表型的对应关系,判断其是否为该CIPA 家系的致病基因。以期发现全新的致病基因突变位点,为CIPA 的预防与诊断提供借鉴。
1 资料与方法
1.1 研究对象
患者男性,17 岁,因反复骨折3 年入院,自出生后全身皮肤无汗,经常出现持续高热,体温增高与外界温度有关,应用抗菌药物及药物降温治疗无效,物理降温有效。自幼感觉障碍、痛觉消失,对各种疼痛刺激均无反应、有自残倾向,多次骨折并发骨髓炎,智力发育迟缓。
1.2 检查方法
1.2.1 查体 体型消瘦,全身皮肤干燥,体表无汗毛,可见多发外伤瘢痕,部分口唇及舌部尖端残缺,背部肩胛区皮肤有潮热感,无腋毛、阴毛。双手掌侧皮肤增厚、结茧,掌指关节处可见皮肤皲裂。十指末端指节缺如、无指甲。下肢不等长,左下肢缩短约3 cm,膝关节囊松弛,右大腿外侧手术切口部分未愈合,部分深筋膜及肌肉组织坏死、深达骨质,可见黄白色脓液,左足踇指末节缺如,左膝抽屉实验:90°及45°均(+)。
1.2.2 实验室检查 肝功能:谷丙转氨酶23 u/L,谷草转氨酶15 u/L,碱性磷酸酶明显升高:353 u/L,血钙降低2.08 mmol/L,胆碱酯酶降低4 737 u/L,血铁降低:4.4 mmol/L,尿常规未见异常,尿酸明显升高990 μmol/L。
1.2.3 温度觉实验 观察患者不能辨别冷热,存在温度觉障碍。
1.2.4 痛觉检查 观察患者全身绝大部分皮肤对疼痛刺激无反应,耳垂、阴囊部分区域痛觉减退。
1.2.5 X 射线检查 右股骨中下段完全骨折移位,骨皮质变薄,骨质疏松(见图1A、B)。切开复位内固定2 周后,骨折端尚未愈合(见图1C)。2 月前左外上髁骨折内固定术后愈合不良(见图1D)。
1.2.6 CIPA 患者皮肤及皮下活组织病理检测 采集CIPA 患者皮肤及皮下组织,进行HE 染色及免疫组织化学检测,观察皮肤是否神经末梢缺如,检测S-100 蛋白表达。
1.3 基因测序
NTRK1 及FAM134B 基因外显子测序:采集患者、其父母及胞弟外周静脉血,提取基因组DNA(使用QIAamp DNA Blood Mini 试剂盒,德国)[15]。使用Primer 3 设计引物,扩增NTRK1 的1~17 外显子及其与内含子交界区。PCR 反应体系共30 μL:Taq mix15 μL,正反向引物各1 μL,基因组DNA 2 μL 和ddH2O 11 μL。PCR 反应条件:95℃预变性5 min,95℃变性30 s,退火30 s;72℃延伸20~60 s,循环35 次;总延伸72℃×10 min。PCR扩增产物纯化后使用ABI3700 荧光自动测序仪(美国赛默飞世尔)进行测序[15],测序结果与参考序列NM002529.3 比对,以确定NTRK1 突变位点及类型。并进一步与NCBI SNP、人类基因突变数据库(http://www.hgmd.cf.ac.uk/ac/index.php)和遗传性周围神经病变突变数据库(http://www.molgen.ua.ac.be/CMTMutations/Mutations/Mutations.cfm)进行比对,排除变异位点为基因多态性位点,并确定其是否为新的突变位点。
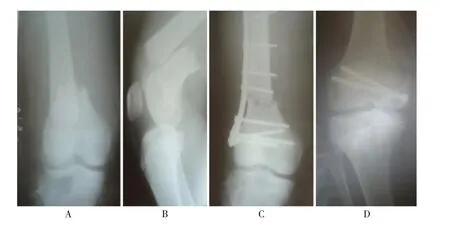
图1 CIPA 患者的X 线表现
2 结果
2.1 常规检查结果
患者病史采集符合典型CIPA 临床表现:该CIPA 患者出生后出现反复持续高热,体温随外界温度增高,全身皮肤无汗。自幼疼痛刺激感觉障碍,痛觉消失,有自残倾向,频繁受伤,多次骨折并发骨髓炎,恢复慢。智力发育不正常,伴有轻度的智力障碍。
CIPA 患者皮肤及皮下组织病理检查:镜下观察表皮未见毛囊,可见汗腺萎缩(见图2、3)。
2.2 基因测序结果
CIPA 家系成员(见图4)基因测序编号父亲(1 号)、母亲(2 号)、患者(3 号)及患者弟弟(4 号)。
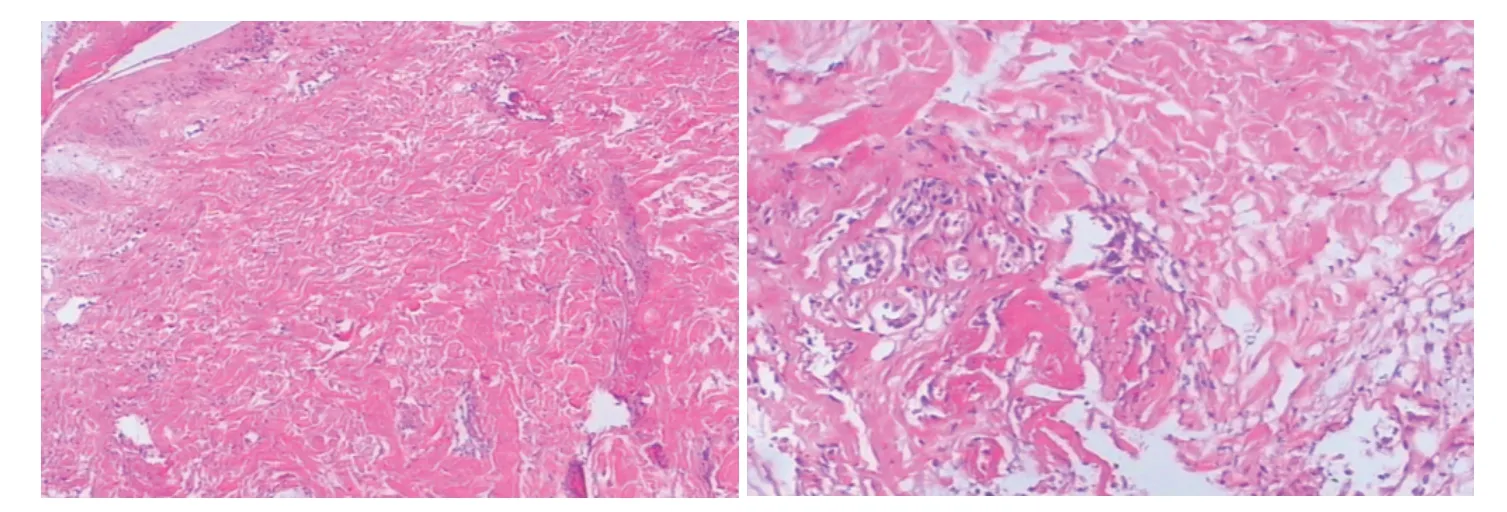
图2 CIPA 患者皮肤的显微镜检查
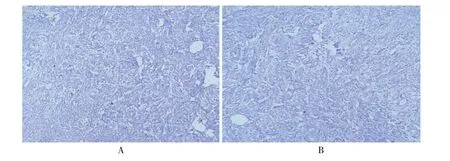
图3 CIPA 患者皮肤免疫组织化学检查
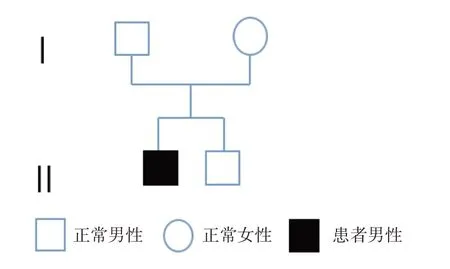
图4 该CIPA 患者的遗传家族图谱
2.3 NTRK1 测序结果
首先对CITP 家系1 号、2 号和4 号个体NTRK 基因1 进行测序,然后与NTRK1 基因参考序列NM_001012331 进行比对,结果发现由于NTRK1 基因的第一个外显子完全位于高GC 区(%GC=70%),以至于用专门扩增高GC 含量的DNA 聚合酶也不能扩增该外显子。对NTRK1 其他外显子的分析结果如下:NTRK1 的外显子3-12、14 和16 均正常。其中在外显子13 有一个常见SNP,rs6334,C/T,1 号患者为C/C 纯合,2 号患者为T/T 纯合,4 号患者为C/T 杂合。外显子2 序列(见图5)在4 个样品中完全正常,然而在内含子2 的GT 后面碱基2 号样品中为G,在1 号、3 号和4 号样品中为一个Indel 突变。
外显子15(见图6)中679 位氨基酸缬氨酸GTG(缬氨酸,V)→ATG(蛋氨酸,M)。1 号样本为G TG;2 号样本为G/A TG;3 号样本为G/A TG;4 号样本为G TG。该突变为新突变,为非同义突变。
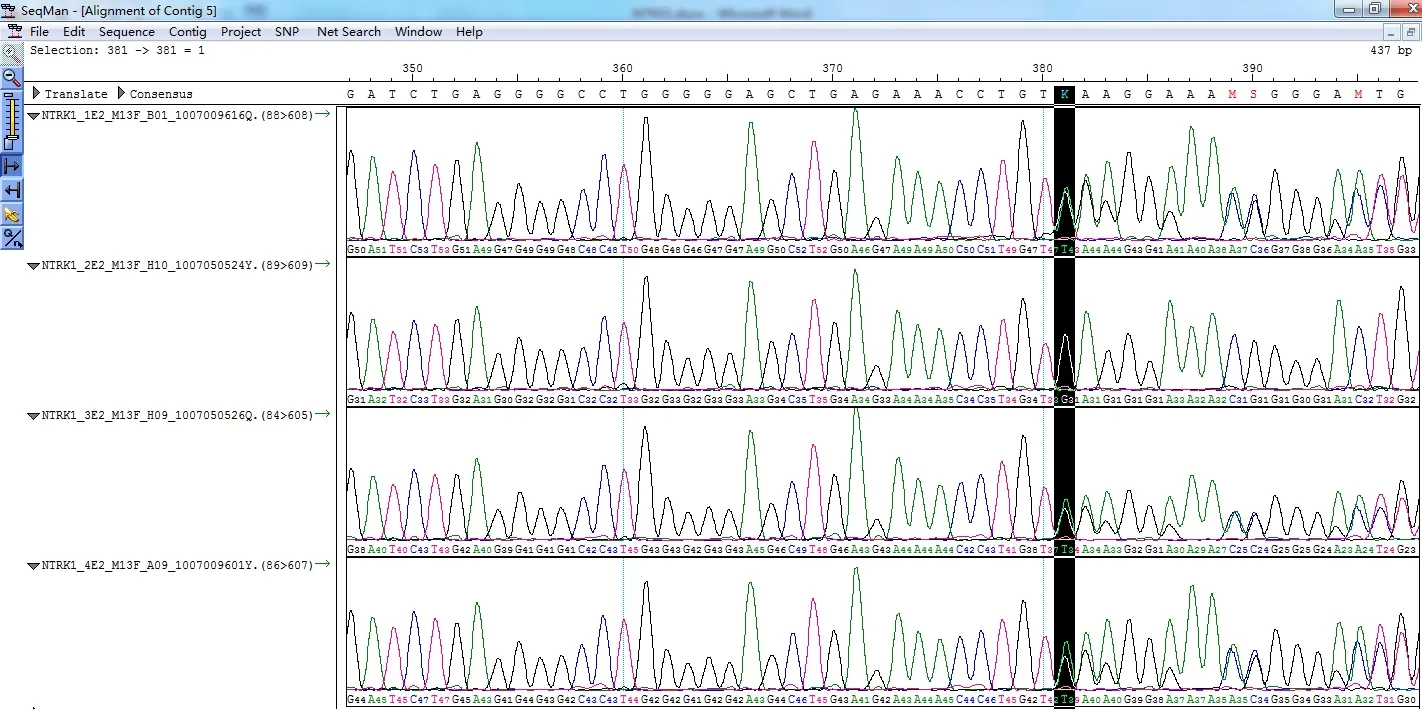
图5 CIPA 家系成员NTRK1 基因中内含子2 的测序结果
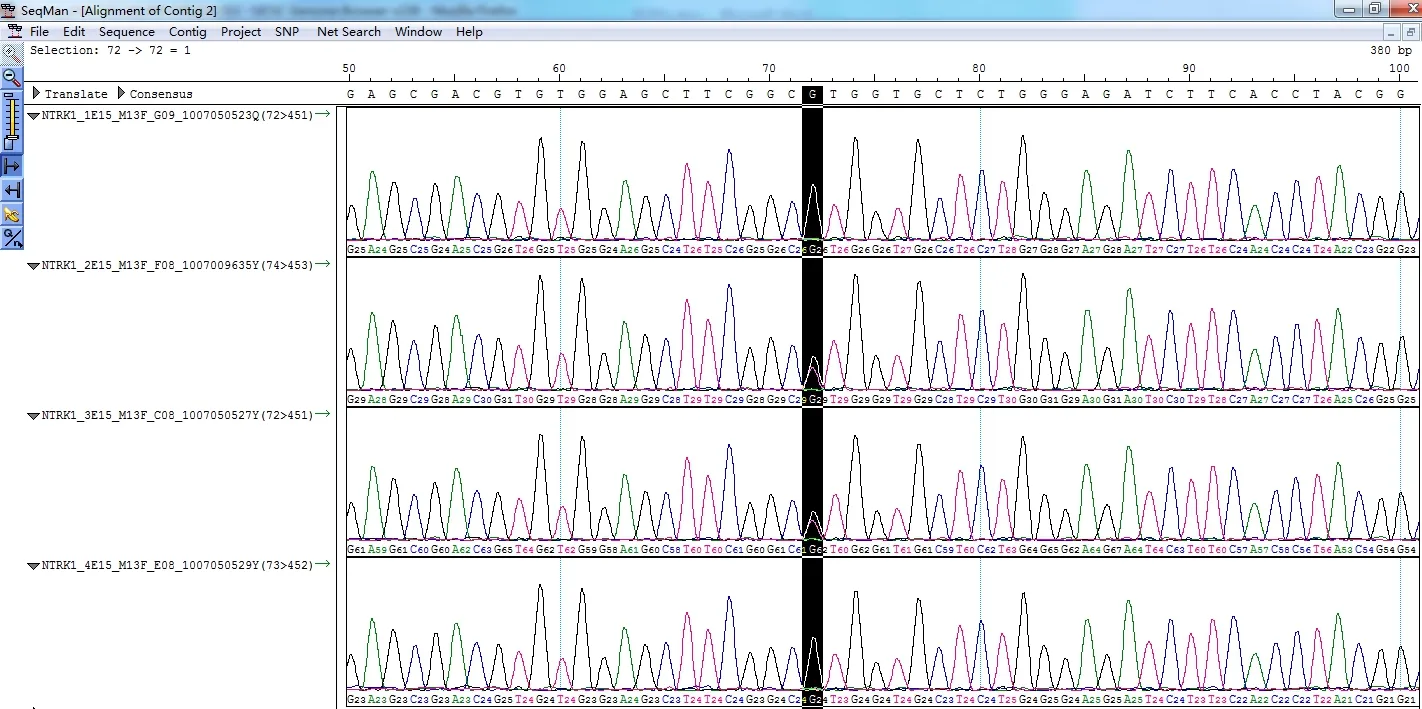
图6 CIPA 家系成员NTRK1 基因中外显子15 的测序结果
其次对CITP 家系的1 号、2 号和4 号个体FAM134B 基因进行测序,然后与FAM134B 基因的参考序列NM_001034850 进行比对,结果发现由于该基因的第一个外显子也位于高GC 含量区域,故该外显子亦未扩增成功;同时因为在该基因内部有较多相对简单序列的重复,故测序成功率较低,虽然进行了多次扩增和测序,目前还有一些外显子未扩增成功或测序效果较差。可考虑进行克隆测序,但会增加实验成本。
3 讨论
CIPA 是一种罕见的常染色体隐性遗传病[1-2]。在国外,CIPA 患者中已有NTRK1 为其主要致病基因报 道[10-13]。INDO 等[14]于1996 年已将NTRK1 基因突变确定为CIPA 的致病原因。NTRK1 蛋白胞外结构域是神经生长因子(nerve growth factor,NGF)的结合位点,可诱导神经轴突生长和促进胚胎感觉和交感神经元存活。NGF 作用通路中的缺陷会影响其他神经元存活,并导致细胞凋亡。其中NTRK1 基因的突变,可影响神经细胞外结构,进而影响细胞内信号的正常传导。NTRKl 基因突变阻断NGF 的作用通路,从而导致疾病的发生。CIPA 患者因为缺少对NGF 信号的转导,造成依赖NGF 的神经元在发育中凋亡,致使背根神经节和皮肤中无细小的有髓鞘神经纤维与无髓鞘神经纤维,皮肤中的汗腺缺乏自主交感神经元的支配,最终造成浅表性痛觉的丧失和无汗现象[7]。该CIPA 患者皮肤及皮下组织病理活检证实汗腺萎缩,皮肤周遭神经无髓鞘,同时存在小有髓鞘纤维缺失,可以解释CIPA 患者痛觉减退和无汗症状。
NGF 除作用神经系统外[8],可参与部分过敏性、免疫缺陷性和自身免疫性疾病等的发病[9]。CIPA 患者由于NTRK1 分子存在缺陷,NGF 信号无法得正常传递,导致中性粒细胞的趋化活性变弱[12]和B 淋巴母细胞系功能失常[13],机体抵抗力差,容易出现感染。
随着外显子技术的发展,对NTRK1 基因的研究更加深入、更加系统[16],尤其是对其基因内多态位点的研究[3]。近年来多项研究发现,CIPA 存在多种NTRK1 基因突变[3,14,17-18]。但突变方式迄今尚未完全阐明,据不完全统计,32 个CIPA 患者及家庭成员的基因分析确定了25 种突变[14]。其中6 个为移码突变,4 个为无义突变,11 个为错义突变,4 个为剪接突变[3]。此后其他突变也被世界各地学者陆续报道[18-22]。截至目前人类基因突变数据库(http://www.hgmd.cf.ac.uk/ac/index.php)已确定的CIPA 患者NTRK1 基因突变总数共79个,分布于NTRK1 整个区域,多数突变位于12~17 外显子区,包括43 个错义突变或无义突变、11个剪接异常突变、12 个微小缺失突变和10 个插入突变,以及仅有1 个大片段缺失突变,其位于gDNA 水平上的985bp,g.7335164_7336545 突变[23]表现为CIPA。其中2/3 的NTRK1 基因突变集中在日本。
该CIPA 家系成员NRK1 基因外显子测序中,外显子2 序列在4 个样品中完全正常,然而在内含子2 的GT 后面碱基在2 号样品中为G,在1号、3 号和4 号样品中为一个Indel 突变,由于该突变在剪接位点内,其产生的后果极为严重,会影响内含子2 或外显子2 的剪接。外显子15 中,发现679 位氨基酸缬氨酸GTG(缬氨酸,V)突变为ATG(蛋氨酸,M),在1 号、4 号样本为G TG;2 号样本为G/A TG;3 号样本为G/A TG。该突变为新突变,为非同义突变,分析该突变只有母亲和患者存在。结合内含子2 中只有母亲不存在Indel 突变,推测内含子2 中Indel 突变和外显子15 中679 位氨基酸缬氨酸GTG(缬氨酸,V)→ATG(蛋氨酸,M)共同作用下,可能会导致该疾病的发病,但因该疾病为罕见病例,样本量有限,进一步验证困难。
有研究发现,HSAN Ⅱ型(先天性感觉性神经病)致病原因为5 号染色体上的FAM134B 基因发生了变异[15]。FAM134B 基因常见于背根节神经元中,后者是负责将感觉信息传递给中枢神经系统的初级感觉神经元。FAM134B 基因变异也会导致其无法表达,使患者痛感丧失或减弱。由于CIPA致病基因迄今未完全阐明,HSAN Ⅱ型及HSAN Ⅳ型(CIPA)均为感觉障碍性疾病,均表现痛觉减退或消失,不能完全排除FAM134B 为CIPA 致病基因。相关文献未查见CIPA 患者行FAM134B 基因检测报道,该CIPA 家系成员FAM134B 基因外显子测序未发现有意义突变,部分外显子测序不成功,需进行克隆测序以进一步明确。
综上所述,通过病史采集、体格检查、痛温度觉实验及皮肤病理活检,可以临床确诊该患者为先天性无痛无汗症。通过外显子测序方法,发现NTRK1 基因内含子2 中Indel 突变和外显子15中679 位氨基酸缬氨酸GTG(缬氨酸,V)→ATG(蛋氨酸,M)突变,推测此两种突变共同作用下,可能会导致该疾病的发病,但进一步验证困难。FAM134B 基因未发现有意义突变,不能验证为致病基因,可能存在其他致病基因,需要进一步研究。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01
中国生殖健康(2020年4期)2021-01-18
山地农业生物学报(2020年2期)2020-11-09
现代农业科技(2020年15期)2020-08-16
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
湖北农业科学(2014年11期)2014-09-10
