
1例以心脏受累为特点的DES基因突变鉴定及表型分析
2021-04-03 06:46:40周源谢利剑肖婷婷徐萌张永为李艳萍
儿科药学杂志 2021年4期
周源,谢利剑,肖婷婷,徐萌,张永为,李艳萍
(上海交通大学附属儿童医院,上海市儿童医院,上海 200062)
DES基因编码的结蛋白(Desmin)是一种在心脏、骨骼肌和平滑肌组织中表达的肌肉特异性中间丝蛋白[1]。Desmin与其他中间丝蛋白相互作用,形成胞浆内网络,维持细胞收缩装置和细胞其他结构元件之间的空间关系,并负责细胞与细胞、细胞与基底膜的连接;Desmin负责肌节的组成、装配和信号传导等,协调机械力的传递,维持肌节收缩的稳定性[2]。DES基因突变呈家族性或散发性,多为常染色体显性或隐性遗传[3]。目前已发现DES基因的各种突变类型包括点突变、插入突变、较小的缺失突变和较大的外显子跳过缺失突变[1]。Desmin突变在1980年由Edstron首次报道[4]。DES基因突变可造成Desmin在肌细胞胞浆内聚集,常与心脏受累和骨骼肌病变有关。超过70%的致病DES基因突变表现为心脏受累,可与任何形式的心肌病相关,主要表现为扩张型心肌病[2,5]或限制型心肌病(dilated cardiomyopathy,RCM)[6-7],也有肥厚型心肌病(hypertrophic cardiomyopathy,HCM)[8]、心律失常性心肌病[9-10]的报道,常伴有传导阻滞,严重者甚至出现心力衰竭和心源性猝死。在扩张型心肌病中DES基因突变的发生率在1%~2%[11]。本研究拟对1例在我院治疗并通过全外显子测序(whole-exome sequencing,WES)提示携带DES基因突变并以心脏受累为主要表现的中国患儿进行Sanger测序,以进一步鉴定基因突变,对突变位点行有害性预测,并对其表型进行相关分析。
1 资料与方法
1.1 研究对象
本研究对象为1例中国汉族12岁的男性患儿及其家系。患儿入院前3个月有暴发性心肌炎、心力衰竭、心源性休克、阿斯综合征、Ⅲ度房室传导阻滞、双下肢静脉血栓、肺炎、肝功能损害、多浆膜腔积液病史,并接受了永久心脏起搏器植入治疗。本次因“安装起搏器术后1个月余,随访发现多浆膜腔积液较前增多,髂外静脉血栓较前扩大”来我院就诊,住院期间完善WES提示DES基因c.1360C>T(p.Arg454Trp)杂合突变,既往无高血压、冠心病及糖尿病史,患儿住院接受了相关药物治疗。出院后定期门诊随访。
1.2 方法
1.2.1 资料收集 收集家系详细临床资料,包括病史采集、体格检查、实验室检查、心电图、超声心动图等检查的结果。本研究经过上海市儿童医院伦理委员会批准(伦理号2019R002-F01),所有研究对象签署知情同意书。
1.2.2 生物信息学分析 在人类基因组变异协会(HGVS)标准基础上与参考基因组NCBI Genome比对,并综合人群频率数据库ExAC、千人基因组(1 000 genomes project)、致病性数据库(ClinVar、OMIM)等进行注释。通过NCBI Homologene对突变位点在不同物种间进行氨基酸序列同源性比对。使用SIFT、Polyphen2、Mutation Taster、Mutation Assessor等软件对候选基因的致病性进行预测,评估突变致病性。
1.2.3 Sanger测序 对患儿及其父母相关突变所在区域进行PCR扩增。在NCBI网站下载DES基因DNA全序列(NM_001927.3,Gene ID:1674),采用Primer5软件设计聚合酶链式反应(PCR)引物,由上海生工生物工程有限公司合成;利用PCR试剂盒(天根生化科技有限公司)扩增。引物序列为F:5’-CTGGGCTGAAGGAAAGGTGTT-3’,R:5’-ATCTCTCTTGCCCCACTAGC-3’。PCR反应条件:96 ℃预变性5 min,95 ℃变性45 s,60 ℃退火45 s,72 ℃延伸60 s,经过30个循环,最后72 ℃延伸7 min。PCR产物纯化、利用ABI 3730XL全自动DNA测序仪上机,采用正向测序,与标准序列对比分析基因序列,以进一步验证突变基因。
2 结果
2.1 患儿临床资料
患儿存在严重的心律失常和心肌病变,没有骨骼肌活动障碍的表现。患儿入院前3个月出现暴发性心肌炎、心力衰竭、心源性休克、阿斯综合征、Ⅲ度房室传导阻滞,在外院治疗并接受了心脏起搏器植入。入我院后完善相关检查,实验室检查提示:N末端脑钠肽(BNP)9 618.19 pg/mL,肌酸激酶同工酶(CK-MB)27 IU/L,肌酸激酶(CK)184 IU/L。心电图(图1)提示:VVI起搏器带动良好,可见房室逆转。超声心动图(图2)提示:双房巨大,心包积液,左心室收缩功能减低(EF 55%),下腔静脉及肝静脉增宽,房间隔缺损(0.23 cm)。胸部超声提示:双侧胸腔少量积液。腹部超声提示:瘀血肝,腹腔积液。腹部大血管超声未见异常。WES结果提示筛查出1个致病基因,为DES(NM_001927.3),患儿携带DES基因 c.1360C>T杂合突变。患儿出院后9个月,我院门诊复查超声心动图提示(图2):双房增大,房间隔缺损(0.2 cm),心包积液较前明显吸收,下腔静脉及肝静脉增宽,左心收缩功能正常(EF 68%)。
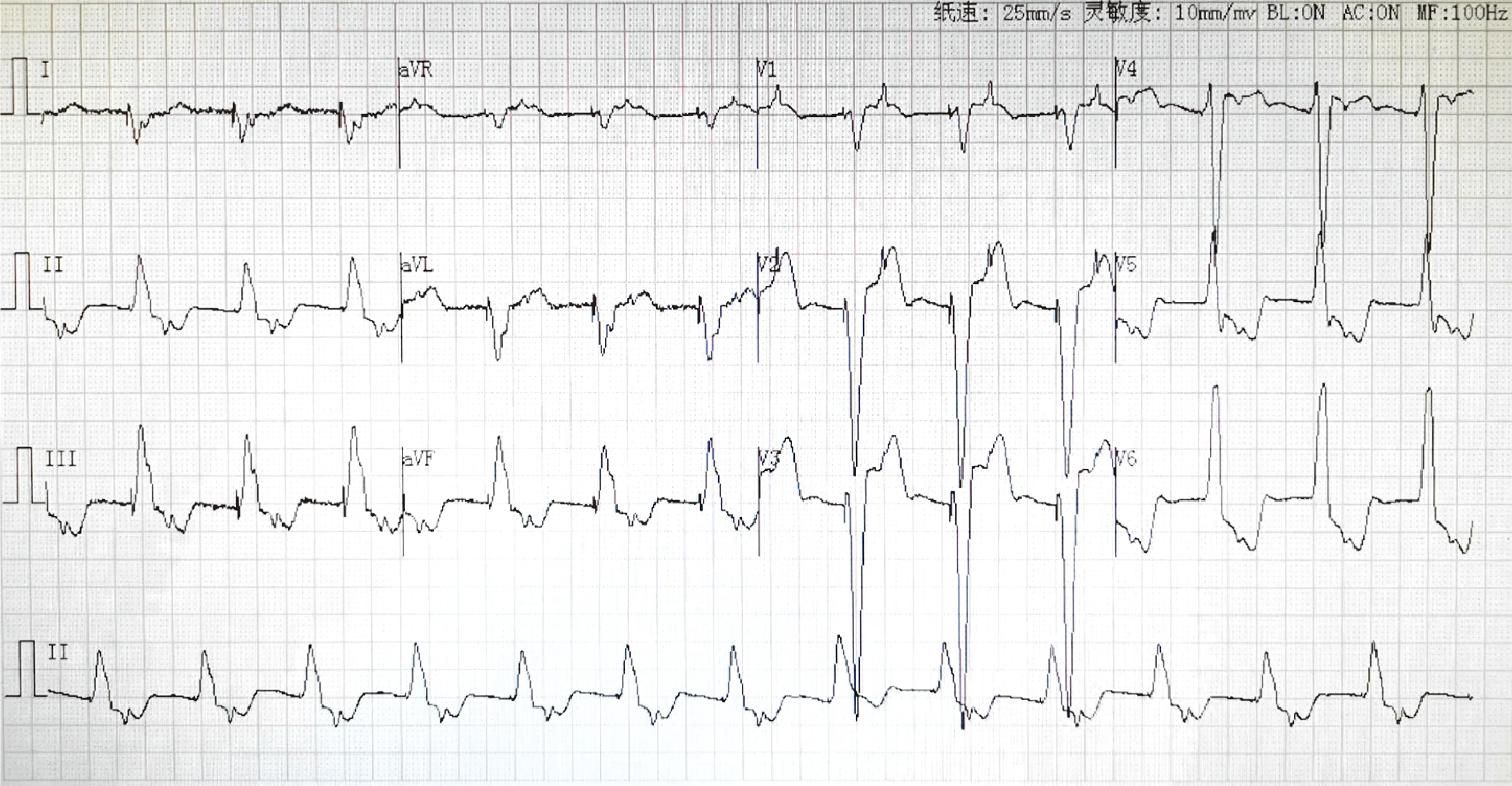
图1 患儿心电图

a~e为入院时检查;f为治疗后9个月复查
2.2 致病基因鉴定
通过Sanger测序进一步证实突变,患儿携带DES基因c.1360C>T杂合突变,并行家系遗传筛查,其父母均无突变,并否认家族心脏病史(家系遗传图见图3)。该突变导致精氨酸突变为色氨酸,通过NCBI Homologene对DES基因454位点氨基酸的对比,发现该位点在不同物种之间有高度保守性(图4)。根据OMIM数据库、ClinVar数据库和临床表型分析,认为该基因突变与患者的临床表型有关联。该患儿携带的DES基因c.1360C>T(p.R454W)突变在ExAC数据库、OMIM数据库、千人基因组数据库均无人群频率报道,ClinVar数据库显示既往有该突变位点报道(表1),临床意义为致病/可能致病。通过PolyPhen2、SIFT、Mutation Taster软件进行DES基因c.1360C>T(p.R454W)突变的有害性预测,结果均提示有害或致病。

注:蓝色代表胞嘧啶脱氧核糖核酸(C);绿色代表腺嘌呤脱氧核糖核酸(A);红色代表胸腺嘧啶脱氧核糖核酸(T);黑色代表鸟嘌呤脱氧核糖核酸(G);双红线间为突变位点,患儿父母均显示蓝色波峰(C),患儿该位点显示红色主峰下出现蓝色套峰

图4 DES基因第454突变位点在不同物种的氨基酸同源性序列比对结果
3 讨论
DES基因位于2q35染色体上,共有9个外显子,在脊椎动物中高度保守。DES基因突变常造成肌肉病变,且具有显著临床异质性。骨骼肌受累表现为肌肉无力,从四肢远端肌肉开始,逐渐扩展到躯干、颈部屈肌、面部。也可出现肠道消化不良、假性肠梗阻等胃肠平滑肌受累症状,严重者可出现呼吸肌受累。患者的发病年龄和病程进展速度差异很大,可在任何年龄出现症状,通常在30岁左右发病[12]。经研究,心肌中的Desmin蛋白比骨骼肌更丰富,是浦肯野纤维的主要组成部分。浦肯野纤维是一种特殊的心肌传导系统,在心脏收缩中起协调作用。超过20%的DES基因突变患者表现为孤立的心脏症状,不涉及骨骼肌[5]。Li Z等[13]用小鼠模型敲除Desmin发现,DES-/-小鼠没有影响发育,但成年小鼠的骨骼肌、平滑肌、心肌出现无组织、肿胀、纤维紊乱的不同形态异常及功能缺陷,说明Desmin对肌肉的分化并非必需,但对组织完整性的维持与增强必不可少。Desmin缺失造成的肌肉结构破坏,包括肌原纤维横向排列的丢失、肌原纤维锚定对肌膜的扰动、线粒体数量和组织的异常、细胞核形状和位置的丢失,最终导致涉及心脏、骨骼和平滑肌的多系统疾病[3]。

表1 DES基因c.1360C>T(p.R454W)突变报道情况及临床表现
Desmin蛋白的分子结构由三部分组成,包括一个α螺旋卷曲杆域的中央区和两侧非螺旋的氨基头端和羧基尾端,中央杆区(1A、1B、2A、2B)被3个短多肽打断[17]。经研究,2B区域的突变可能在Desmin二聚体之间的螺旋内和螺旋间的离子桥上产生一个关键的断裂,因改变了Desmin二聚体和低聚体组装及其与膜蛋白的连接而致病,也是报道突变最多的区域[9]。1A区域突变不影响纤维网络的形成,但可造成功能障碍。1B区域突变较为罕见[2]。非螺旋尾区缺乏七肽重复模式,主要参与Desmin与其他细胞骨架蛋白的相互作用,建立胞质中间丝网络[9]。大部分尾部区域的突变看似没有破坏细胞骨架结蛋白网络的形成,但足以造成功能障碍[11]。头部和尾部区突变主要发生在孤立的心脏病表型患者中[12]。
本研究发现1例位于DES基因尾部区域突变的患者,表现出严重的心脏表型。本例患儿在住院期间通过WES检查发现携带DES基因c.1360C>T(p.Arg454Trp)杂合突变。该位点位于DES基因尾部的8号外显子上。为证实该突变,进一步行Sanger测序,证实了DES基因第1 360位的胞嘧啶突变为胸腺嘧啶,导致第454位的精氨酸(Arg,R)被色氨酸(Trp,W)取代。通过对患儿父母的遗传筛查均未发现检测出突变基因,故本例患儿为新发变异,属散发性病例。这也不能够排除后天环境因素的影响。通过氨基酸同源性序列比对发现,DES基因的第454位氨基酸残基在不同物种间存在高度保守性。查询ExAC数据库、OMIM数据库、千人基因组数据库均无人群频率报道,说明该突变罕见。ClinVar数据库显示既往有该位点突变报道,突变意义为致病或可能致病。经过PolyPhen2、SIFT、Mutation Taster等软件预测显示该位点突变很可能是有害变异,说明c.1360C>T突变很可能会影响DES基因表达和蛋白质的功能。本例患儿主要表现为双心房巨大、心包积液、左心室收缩功能减低(EF 55%),既往心电图提示存在Ⅲ度房室传导阻滞,并接受了永久起搏器植入。实验室检查提示该患儿 CK-MB 轻度升高,CK属正常范围,因目前暂未出现活动障碍、骨骼肌异常和远端肌力改变等表现,故住院期间未进行肌肉活检、肌电图检查。因患儿存在心功能异常,BNP异常升高,同时因存在多浆膜腔积液、双下肢静脉血栓形成和支气管肺炎,在住院期间予美罗培南抗感染、磷酸肌酸钠联合辅酶Q10营养心肌、复方甘草酸苷(美能)护肝、呋塞米联合螺内酯利尿、阿司匹林抗血小板聚集等对症综合治疗,并完善基因检测。经过治疗,患儿病情可,无不适后予出院。出院后继续抗凝、利尿、保心、保肝等对症治疗,定期门诊随访及复查,9个月后复查心脏B超提示心包积液明显减少,左心收缩功能已达正常。
既往已有DES基因c.1360C>T(p.Arg454Trp)突变的相关病例报道与机制研究。2007年,DESp.R454W突变首次在1例因肥厚型心肌病(HCM)而出现运动不耐受和呼吸急促的15岁男性患者身上被发现,该患者同时携带了条件致病性的Tilin p.Q74K基因突变,最后接受了心脏移植。通过体外组装方案发现,当突变体与野生型DESmin蛋白共配时,形成的混合纤维存在固有的不亲和性[14]。Otten E等[10]在以房室传导阻滞(atrioven-tricular block,AVB)为主要表现的严重双心室心肌病、不伴有骨骼肌表现的一对兄妹及其父亲身上也发现了DESp.R454W突变,均在20岁左右接受了心脏起搏器植入,但因进行性的呼吸困难和心力衰竭,患者均在30岁前后出现死亡。Vattemi G等[15]对21例肌原纤维性肌病的患者进行临床、形态学和遗传学研究,发现其中17例出现肌无力表现,接受心脏评估的10例患者中,7例出现了心脏表型异常。经基因诊断发现,其中有一对兄弟分别携带DES基因p.S7F和p.R454W突变。Bär H等[18]进行震荡剪切试验和高频挤压流测量研究发现,R454W突变不同于尾部其他位点突变,R454W不能形成正常大小的纤维网络,而尾部其他突变体(如T442I、K449T、S460I、I451M)与野生型Desmin相比,纤维网络大小和纤维硬度值在相同范围,只是在纤维应变张力上出现了明显降低,造成非仿射网络形变,表明DES基因尾部突变影响纤维内部结构和纤维间相互吸引作用,对纤维应变张力产生影响。近年,Weihl C C等[16]在一家系中发现,DESp.R454W位点突变,先证者表现为四肢远端肌力减弱,心脏射血分数减低(EF 40%),双房肥大和传导阻滞。通过对患者肌肉组织活检发现Desmin的内含物与自噬空泡病理相同,提出Desmin蛋白的集聚可能激活了自噬系统,自噬功能及功能障碍可能是造成其发病的重要介质,也可能是一个治疗靶点。此外,Brodehl A等[5]在一家系中发现,携带DESp.L136P突变的先证者有心脏表型,该突变所致的临床表现也未累及骨骼肌,并提出这可能是由于受损的骨骼肌中存在卫星细胞,帮助了骨骼肌的更新修复,而哺乳动物心脏组织再生能力较低,以及心肌细胞与骨骼肌细胞间的结构差异,故导致DES突变对心肌和骨骼肌产生了不同程度的影响。JurcuR O等[19]报道了1例携带DESc.1297C>A p.(Pro433Thr)突变的25岁女性患者,临床特征为伴有轻度骨骼肌病变的严重限制型心肌病。该突变位点在基因中的位置与本研究位点相近,都属于DES基因尾部突变。患者从20岁起有反复晕厥病史,先后出现Ⅲ度房室传导阻滞、限制型心肌病(RCM)的心脏受累表现,接受了起搏器治疗,在初次出现心脏表型的5年后才出现轻度肌无力的骨骼肌受累,五年间检测指标发现肌酸激酶(CK)轻度进行性升高。
结合本例和既往相关报道可见,DESc.1360C>T(p.R454W)突变造成的心脏表型存在临床异质性,表型通常较严重,发病年龄较早,心脏不良事件发生率高,预后差,部分病例可不伴有骨骼肌的受累。在既往报道中,DESp.R454W突变多为家族遗传。本例否认相关家族史,其父母通过WES未检测出携带相同突变,因此出现了偶发性突变。且本例患儿发病年龄为12岁,早于既往已报道的病例。
DES基因突变可导致AVB,大多数出现AVB的患者同时合并临床表现明显的肌病,但也有仅表现为RCM、AVB但无周围肌损害症状的DES突变患者的病例报道,但Ⅲ度房室传导阻滞也可能是疾病的最初表现[20-21]。本例患儿存在Ⅲ度房室传导阻滞,在外院接受起搏器植入前确诊过暴发性心肌炎,故存在暴发性心肌炎加重了DES基因突变所致AVB及其他心肌病变程度的可能,目前暂无DES基因突变与暴发性心肌炎的相关研究报道。此外,该患儿仅出现心肌受累表现,可能处于疾病进展的初期,心脏表型先于肌病出现的可能性不能排除。在经过保心、利尿、抗凝等对症治疗后,复查心脏彩超提示心包积液吸收明显,左心收缩功能达到正常,但双心房的扩张并没有显著改善,可能存在心肌重构,已造成不可逆的损害。至于将来是否会出现病情进展或伴有骨骼肌病变,还需长期随访观察,定期监测BNP、CK、CK-MB等指标变化。
总之,DES基因突变所致的疾病以表型各异的心肌病变为主要表现之一。当存在严重心律失常、心肌病表现时,应建议患者完善基因检测,明确病因、遗传方式及遗传因素,有助于早期干预和治疗,延缓疾病进展,同时对相关家系成员进行早期筛查,告知注意相关疾病的预防,对突变携带者起到预警作用,定期检查以早期识别疾病,也可给予家族遗传咨询,做到风险规避。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国生殖健康(2020年2期)2021-01-18 02:51:26
中国临床医学影像杂志(2019年1期)2019-04-25 06:49:44
小学生导刊(2018年13期)2018-06-29 03:49:00
中国中医药信息杂志(2016年5期)2016-12-01 06:07:21
中国运动医学杂志(2016年3期)2016-07-10 12:07:23
中国运动医学杂志(2016年3期)2016-07-10 12:07:23
医学研究杂志(2015年5期)2015-06-10 06:43:26
国际心血管病杂志(2015年5期)2015-02-27 12:11:33
河南医学研究(2014年5期)2014-02-27 14:52:41
