
1例复合杂合突变所致的凝血因子Ⅻ缺乏症家系分析*
2021-04-01 09:49邹安庆王明山金艳慧夏虹刘斯奇陈怡
临床检验杂志 2021年2期
邹安庆,王明山,金艳慧,夏虹,刘斯奇,陈怡
(温州医科大学附属第一医院a.医学检验中心,b.血液科,浙江温州325000)
凝血因子Ⅻ(coagulation factor Ⅻ,FⅫ)是一种单链糖蛋白,由肝细胞产生,呈现为一种非活性丝氨酸蛋白酶前体分泌到血浆,相对分子质量(Mr)为80 000,由596个氨基酸残基组成[1]。FⅫ可激活内源凝血途径,并参与纤溶途径的激活。先天性FⅫ缺乏症是一种常染色体隐性遗传病,其病因常为位于染色体5q33-qter上的FⅫ基因突变[2]。其可分为3类:交叉反应物质阴性(CRM-)型(FⅫ:Ag不能被检测)、阳性(CRM+)型(FⅫ:Ag正常)和降低(CRMred)型(FⅫ:Ag降低)。然而,有些FⅫ缺乏症患者出血表现轻微,有些患者可发生静、动脉血栓[3-5]。FⅫ基因的46T/T型在肺血栓患者中比例更高,而凝血因子Ⅻ活性更低[6]。本研究报告1例FⅫ缺乏症患者,分析先证者与其家系的表型与基因型,同时利用生物信息学软件分析突变的可能危害,初步探讨其分子发病机制。
1 对象和方法
1.1对象 先证者,男性,54岁,温州人,吸烟35年,2020年3月26日因“胸闷咳嗽1月余”到温州医科大学附属第一医院就诊,拟“肺占位损伤”收治入院。入院后完善相关检查,CT示“右肺中央型肺癌伴中下叶阻塞性不张”,气管镜检查见右总支气管、气管下端新生物,病理提示鳞癌及鳞状上皮重度不典型增生伴癌变。常规凝血功能检查发现活化部分凝血活酶时间(activated partial thromboplastin time,APTT)179.2 s,查凝血因子发现FⅫ活性(coagulant factor Ⅻ activity,FⅫ:C)为1%,FⅫ抗原(coagulant factor Ⅻ antigen,FⅫ:Ag)含量为1%。先证者肝肾功能检查结果正常,平时无明显自发出血或血栓形成症状,诊断为FⅫ缺乏症。先证者及其家系(共3代6人)参与本研究,包括先证者及其父亲、妻子、长子、女儿和次子。家系成员均无自发出血倾向,也无血栓形成史,否认近亲婚配。家系遗传图谱见图1。
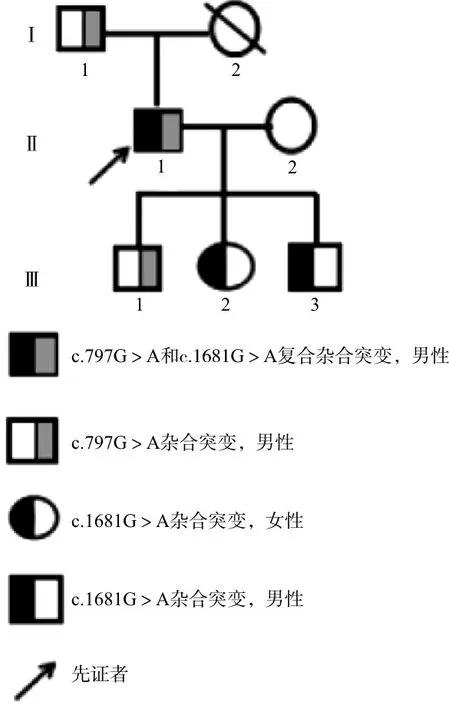
图1 遗传性FⅫ缺乏症家系图
100名体检健康者作为正常对照,男53名、女47名,平均年龄32岁(年龄范围16~60岁),均无肝肾疾病,无血栓或出血史。
所有研究对象均签署知情同意书。本研究得到温州医科大学附属第一医院伦理委员会的批准(批准文号:2012-17)。
1.2标本采集与处理 采集先证者及家系成员外周静脉血2.7 mL于含0.109 mol/L枸橼酸钠的真空采血管中,3 000 r/min离心10 min,上层血浆用于凝血指标检测,下层血细胞用于提取基因组DNA和PCR扩增。
1.3主要仪器与试剂 STA-Max全自动血凝仪(法国Stago公司),ABI 2720 PCR扩增仪(美国Applied Biosystems公司),ABI PRISM 3700测序仪(美国Applied Biosystems公司);PT、APTT和FⅫ:C等凝血试剂(法国Stago公司);FⅫ:Ag检测试剂盒(上海西唐生物科技公司);DNA提取试剂盒(北京天根生化科技公司)。
1.4方法
1.4.1血凝功能检测 凝血功能试验,包括APTT、PT和FⅫ:C,均采用一期凝固法测定。采用ELISA法测定FⅫ:Ag。
1.4.2DNA抽提和PCR扩增 按试剂说明书抽提先证者及家系成员外周血基因组DNA。根据NCBI中的FⅫ基因序列(GenBank登录号:AF538691),应用Primer 5软件共设计7对引物以覆盖FⅫ基因的14个外显子区域及其侧翼序列,引物序列见表1。由上海桑尼生物科技公司合成。
PCR扩增:反应体积共25 μL,包括Taq PCR Master mix 12.5 μL,ddH2O 7.5 μL,DNA模板3 μL,正向引物1 μL,反向引物1 μL。反应条件:95 ℃预变性5 min,95 ℃变性30 s,根据不同引物分别以相应退火温度(54~60 ℃)退火30 s,72 ℃延伸45 s(共35个循环),72 ℃延伸10 min。PCR产物采用Goldview标记,在15 g/L琼脂糖凝胶上进行电泳,经电泳检测合格的PCR产物送上海桑尼生物科技公司测序。测序结果用Chromas软件与美国NCBI基因库所公布的FⅫ基因序列(GenBank登录号:AF538691.1)进行比对,寻找基因突变位点,发现错义突变位点则通过反向测序予以证实。先扩增先证者FⅫ各外显子及侧翼序列和5′、3′端非翻译区,发现突变位点后再扩增其他家系成员相应外显子片段。

表1 FⅫ基因PCR扩增引物序列
1.4.3突变位点氨基酸保守性及危害性分析 用ClustalX-2.1-win软件将突变氨基酸(Cys247和Gly542)与其7种同源物种(小家鼠,褐家鼠,黑猩猩,猕猴,家犬,牛和热带爪蟾)的氨基酸序列进行比对,分析突变氨基酸的保守性。同源物种氨基酸序列来源:http://www.ncbi.nlm.nih.gov/homologene。
通过4个在线生物信息学软件:PolyPhen-2(http:∥genetics.bwh.harvard.edu.pph2),PROVEAN(http:∥provean.jcvi.org/seq_submit.php),SIFT(http:∥sift.jcvi.org),MutationTaster(http:∥www.mutationtaster.org)(蛋白质参考ID:P00748和蛋白质参考转录本ID:ENST0000025 3496),用评分结果预测突变对蛋白质功能的影响。
2 结果
2.1凝血指标检测结果 家系成员表型检测显示先证者父亲、长子、女儿和次子的FⅫ:C和FⅫ:Ag减少,FⅫ:C分别为43%、45%、53%和51%;FⅫ:Ag分别为48%、43%、54%和57%。APTT、PT和Fib测定结果显示,父亲、长子和女儿的APTT较对照组延长。其他指标未见异常。家系成员的表型和基因型见表2。
2.2FⅫ基因分析结果 先证者FⅫ基因DNA测序发现,先证者第1外显子启动子区46C/T位点多态性为46T/T型、第9外显子存在c.797G>A杂合错义突变(导致p.Cys247Tyr)、第14外显子存在c.1681G>A杂合错义突变(导致p.Gly542Ser)。先证者父亲和长子存在c.797G>A突变杂合子,女儿和次子存在c.1681G>A突变杂合子,妻子为野生型。见表2和图2。
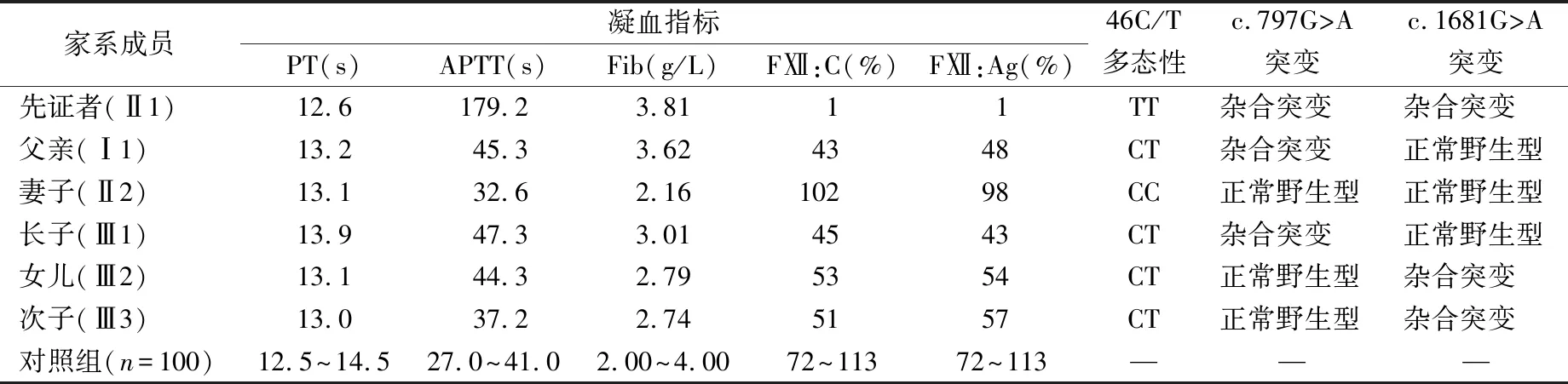
表2 FⅫ缺乏症家系表型及基因型检测结果
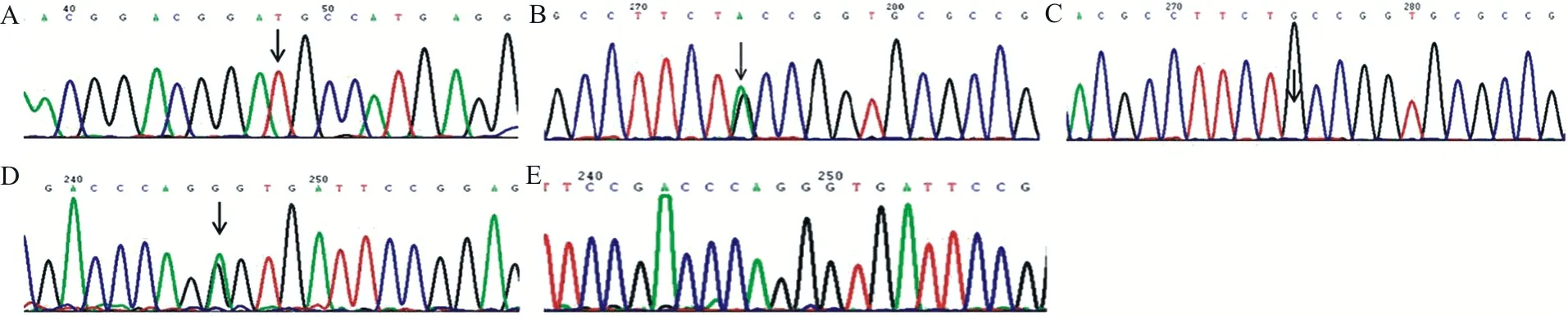
注:A,第1外显子启动子区46T/T型;B,第9外显子c.797G>A杂合突变型;C,第9外显子c.797G>A野生型;D,第14外显子c.1681G>A杂合突变型;E,第14外显子c.1681G>A野生型。
2.3突变氨基酸保守性及危害性分析结果 人类FⅫ基因突变位点氨基酸与NCBI数据库提供的其他7个同源性物种基因氨基酸进行多重比对,结果显示Cys247和Gly542这2个氨基酸在这7个物种间高度保守,见图3和图4。

图3 Cys247保守性分析结果

图4 Gly542保守性分析结果
4个生物信息学软件分析结果表明,c.797G>A错义突变的预测结果为“有害”和“致病性”;c.1681G>A错义突变的预测结果为“可能有害的”、“有害”、“影响蛋白质功能”和“致病性”。见表3。
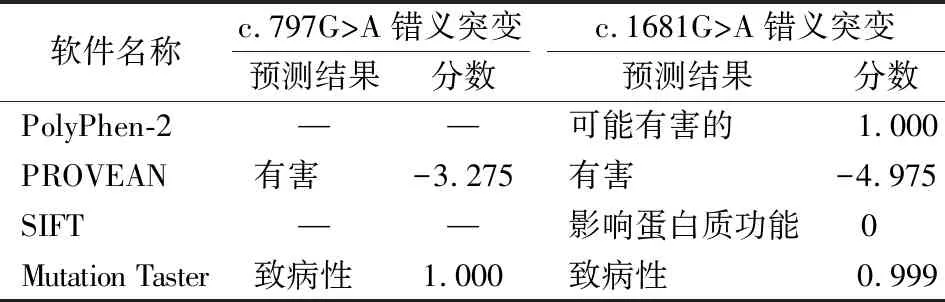
表3 生物信息学软件对c.797G>A和c.1681G>A错义突变的预测结果
3 讨论
FⅫ由596个氨基酸(AA)组成,可分为数个结构/功能区[7],自氨基末端起分别是Ⅱ型纤维连接蛋白区(AA1-88)、表皮生长因子区(AA94-131)、Ⅰ型纤维连接蛋白区(AA133-173)、表皮生长因子区(AA174-210)、Kringle区(AA215-295)、富脯氨酸区(AA296-349)和催化区(AA354-596)。其中催化区是发挥酶解功能的关键,包含激活区和丝氨酸蛋白酶区,活性部分由组氨酸(393)、天冬氨酸(442)和丝氨酸(544)残基组成。
本家系研究发现,先证者的FⅫ基因上存在3个遗传变异,包括第1外显子46T/T位点多态性和2个错义突变(第9外显子存在c.797G>A杂合错义突变导致p.Cys247Tyr和第14外显子存在c.1681G>A杂合错义突变导致p.Gly542Ser)。查询相关文献,Kanaji等[8]于1998年首次报道了第1外显子启动子区46C/T位点多态性。c.1681G>A杂合突变也已有报道[9]。而c.797G>A杂合突变为尚未见报道过的新突变。根据家系研究结果,先证者FⅫ:C和FⅫ:Ag显著减少并接近0,表明突变属于CRM-型。先证者父亲、长子、女儿和次子的突变属于CRMred型,因为FⅫ:Ag分别下降为48%、43%、54%和57%。
c.797G>A和c.1681G>A这2个突变位点分别位于FⅫ的Kringle区和催化区。相关文献报道,c.1681G>A(p.Gly542Ser)位点位在活性位点Ser544附近,突变后改变了氨基酸的极性,影响催化位点的细微空间构象并最终影响FⅫ活性。体外瞬时表达亦显示c.1681G>A突变导致FⅫ变异蛋白合成减少,并伴有分泌障碍[10-11]。
c.797G>A是一个新发现的突变,导致p.Cys247Tyr。FⅫ蛋白的Kringle结构域包含215~295个氨基酸残基,在增强激肽释放酶裂解敏感性方面起到一定作用[12]。在Kringle结构域中已经报道过这几个突变:p.W222G、p.G240E和p.R248G[12-13]。p.G240E和p.R248G的体外表达实验研究表明,这2种突变在高尔基体前室发生了广泛的细胞内降解,导致分泌不足。p.Cys247Tyr与p.G240E/p.R248G相邻,推测p.Cys247Tyr对FⅫ的致病机制与p.G240E/p.R248G相似。为了进一步证明p.Cys247Tyr的致病性,本研究分析了其保守性和可能对蛋白质的影响。在同源物种分析中,Cys247是一个高度保守的位点,被认为是FⅫ的重要功能位点。在线生物信息学软件预测结果为“有害突变”和“致病”,可对蛋白质功能造成一定影响,并引起相关疾病。由于FⅫ的Kringle结构域没有X射线三维结构图,因此无法分析突变蛋白的空间结构。当Cys247被Tyr247取代后,Cys247-Cys271位点间的二硫键消失,此外,p.Cys247Tyr在侧链中引入了一个大的酪氨酸芳环,这可能会形成其他分子间作用力,如氢键和空间位阻等。这些都可能影响蛋白质的正常结构,形成不稳定的蛋白质,更容易被降解,从而引起了FⅫ水平降低。
除c.797G>A和c.1681G>A突变外,本研究中先证者第1外显子启动子区存在46C/T多态性。C>T取代后,在起始密码子上游的4个碱基处形成新的起始密码子,导致起始密码子下游第7个碱基处形成终止密码子,翻译提前终止。先证者妻子的FⅫ:C约为先证者的3倍,这与文献报道的结论相一致[14]。
到目前为止,人类基因突变数据库(HGMD)中已经登记了58个FⅫ突变,Kringle域的突变很少。本文首次报道的c.797G>A丰富了数据库。我们发现先证者APTT的显著延长(179.2s)以及FⅫ活性的显著减低(FⅫ:C为1%)可能是由46T/T、c.797G>A和c.1681G>A复合杂合突变共同引起,导致FⅫ缺乏的发生。
猜你喜欢
当代医药论丛(2022年17期)2022-10-09
临床输血与检验(2022年3期)2022-06-22
实用肝脏病杂志(2022年2期)2022-03-21
今日农业(2021年16期)2021-11-26
种子(2021年3期)2021-04-12
大自然探索(2020年5期)2020-06-19
诊断学(理论与实践)(2020年1期)2020-04-28
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
