
利用N糖苷酶F对单克隆抗体N糖酶解条件的优化
2021-03-30 03:16张博慧贾戴辉程倩许俊彦邵喆黄应峰
生物技术进展 2021年2期
张博慧, 贾戴辉, 程倩, 许俊彦, 邵喆, 黄应峰
宝船生物医药科技(上海)有限公司药物分析部门, 上海 201203
N糖基化修饰是单克隆抗体药物普遍存在的翻译后修饰现象。目前研究表明,糖基化修饰与单克隆抗体药物的功能、药物代谢、免疫原性等关系密切[1-3],如核心岩藻糖缺失能显著增强ADCC效应[4],半乳糖能够增强CDC活性[5];末端N-乙酰葡萄糖影响抗体药物半衰期[6-7];唾液酸化糖型具有抗炎症功能[8];α(1-3)半乳糖和NGNA型唾液酸易引起免疫原性[9]等。N糖对抗体结构也具有重要作用,能稳定CH2结构域,去糖抗体稳定性会变差,更易发生去折叠和聚集[10]。
N糖类型和含量受细胞株、培养条件、纯化和储存[11-13]等过程的影响,因此在单抗药物细胞筛选、工艺优化、纯化、产品放行和稳定性考察中控制和监测N糖含量,保证单抗药物安全性、有效性等尤为重要。目前检测N糖的方法主要有亲水色谱-荧光法[14]、CE-LIF法[15-16]和质谱法[17-18]等。其中亲水色谱-荧光法是目前应用最为广泛的方法,该方法通常需要用糖苷酶水解糖蛋白上的N糖链,再用标记试剂进行衍生化,采用亲水色谱柱分离,荧光检测器检测。由于PNGase F几乎能水解所有哺乳动物细胞产生的N糖,因此得到广泛应用[19]。传统的N糖谱检测方法一般耗时较长,目前快速N糖样品制备的试剂盒已经商品化,大幅提高了N糖的检测效率,但是通常价格昂贵。而应用快速PNGase F酶与传统制备过程相组合成为提高检测效率和降低检测成本的一种选择。本研究基于亲水色谱-荧光法,针对PNGase F水解条件进行了优化,发现和解决了酶解过程中易对结果产生影响的一些因素,建立了高效准确的N糖检测方法,以期为N糖的检测研究提供参考。
1 材料与方法
1.1 实验材料
1.1.1供试品 本研究所用mAb1、mAb2和mAb2-F均为宝船生物医药科技(上海)有限公司生产的IgG1型单克隆抗体。
1.1.2试剂和耗材 N糖苷酶F(PNGase F)、快速PNGase F酶(rapid PNGase F)及变性缓冲液(5×)均购自New England BioLabs公司;无水乙醇、甲酸铵、乙酸、二甲基亚砜(DMSO)、2-氨基苯甲酰胺(2-AB)、氰基硼氢化钠、β-巯基乙醇等均购自Sigma公司;甲酸和乙腈购自Fisher公司;PBS Buffer(1×)购自上海生工;10 kD超滤离心管购自Merck公司;100 mmol·L-1Tris-HCl(含1% SDS,pH 9.0)溶液和非涂层-熔融石英毛细管购自Beckman公司;纯化柱(GlycoCleanTMS Cartridges)购自Prozyme公司;色谱柱ACQUITY UPLC Glycan BEH Amide Column(1.7 μm,2.1 mm×150 mm)购自Waters公司;色谱柱AdvanceBio Glycan Mapping Column(2.7 μm,4.6 mm×150 mm)购自Agilent公司。
1.1.3主要仪器设备 高效液相系统(HPLC,1260)购自Agilent公司;超高效液相系统(UPLC,H-Class Plus)购自美国Waters公司;液质联用系统(LC-MS,Q Exactive)和数据处理软件(Biopharma Finder)均购自美国Thermo公司;毛细管电泳仪(CE,PA800 Plus)购自Beckman公司;真空离心浓缩干燥仪(RVC 2-25)购自德国Christ公司。
1.2 实验方法
1.2.1蛋白沉淀和N糖衍生化 向酶解后的样品中加入3倍体积的冰乙醇,-15~-25℃放置约1 h,高速离心10 min,取上清干燥。加入10 μL 2-AB衍生化试剂,65℃避光孵育3 h。用纯化柱纯化后干燥,50%乙腈复溶,准备上机分析。
1.2.2HPLC色谱条件 仪器采用高效液相系统(Agilent,1260);色谱柱采用Advance Bio Glycan Mapping Column;流动相A为100 mmol·L-1甲酸铵(pH 4.5),流动相B为100%乙腈;进样量2 μL;流速0.5 mL·min-1;荧光检测器激发波长260 nm,发射波长430 nm;色谱柱温度55℃;梯度洗脱。
1.2.3UPLC色谱条件 仪器采用超高效液相系统(Waters,H-class plus),色谱柱采用ACQUITY UPLC Glycan BEH Amide Column。流动相A为100 mmol·L-1甲酸铵(pH 4.5),流动相B为100%乙腈,进样量2 μL,流速0.5 mL·min-1,荧光检测器激发波长330 nm,发射波长420 nm,色谱柱温度60℃,梯度洗脱。
1.2.4质谱参数 应用液质联用系统(LC-MS,Q Exactive)的HESI正离子模式(HESI+),离子源的温度和电压分别为320℃和3.8 kV,离子传输管温度为200℃,其他仪器参数设置均经过优化以获得最佳信号响应;母离子扫描范围700~3 000 m·z-1,分离度15 000,分析时长60 min;数据处理软件采用Biopharma Finder。
1.2.5还原毛细管凝胶电泳(R CE-SDS) 将蛋白沉淀用100 mmol·L-1Tris-HCl(含1% SDS,pH 9.0)溶液复溶,加入β-巯基乙醇还原。仪器采用毛细管电泳仪(Beckman,PA800 plus),毛细管采用非涂层-熔融石英毛细管,PDA检测器,波长220 nm,电动进样(-5 kV)20 s,电压分离(-15 kV)40 min。
2 结果与分析
2.1 缓冲液pH对结果的影响
分别将mAb1和mAb2样品调pH至4、5、6和7,取200 μg进行PNGase F酶解24 h,检测N糖含量。实验结果如表1和图1所示,mAb2在不同pH缓冲液酶解的N糖含量高度一致,说明pH对该样品N糖水解无影响;而mAb1中N糖含量随pH的变化出现明显差异,其中缓冲液pH为4时,峰2(p2)含量出现大幅度升高,相应的峰3(p3)含量明显下降,pH 6和7条件下N糖含量基本一致。
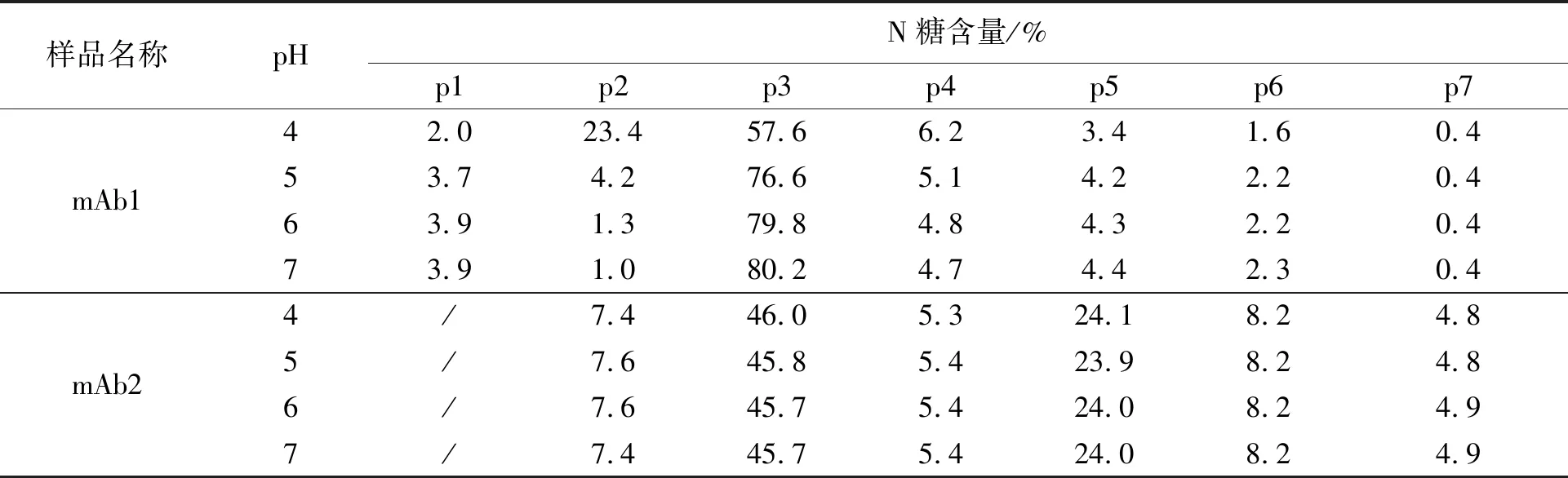
表1 mAb1和mAb2在不同pH缓冲液中酶解的N糖含量Table 1 N-glycan content of mAb1 and mAb2 hydrolyzed in different pH buffer
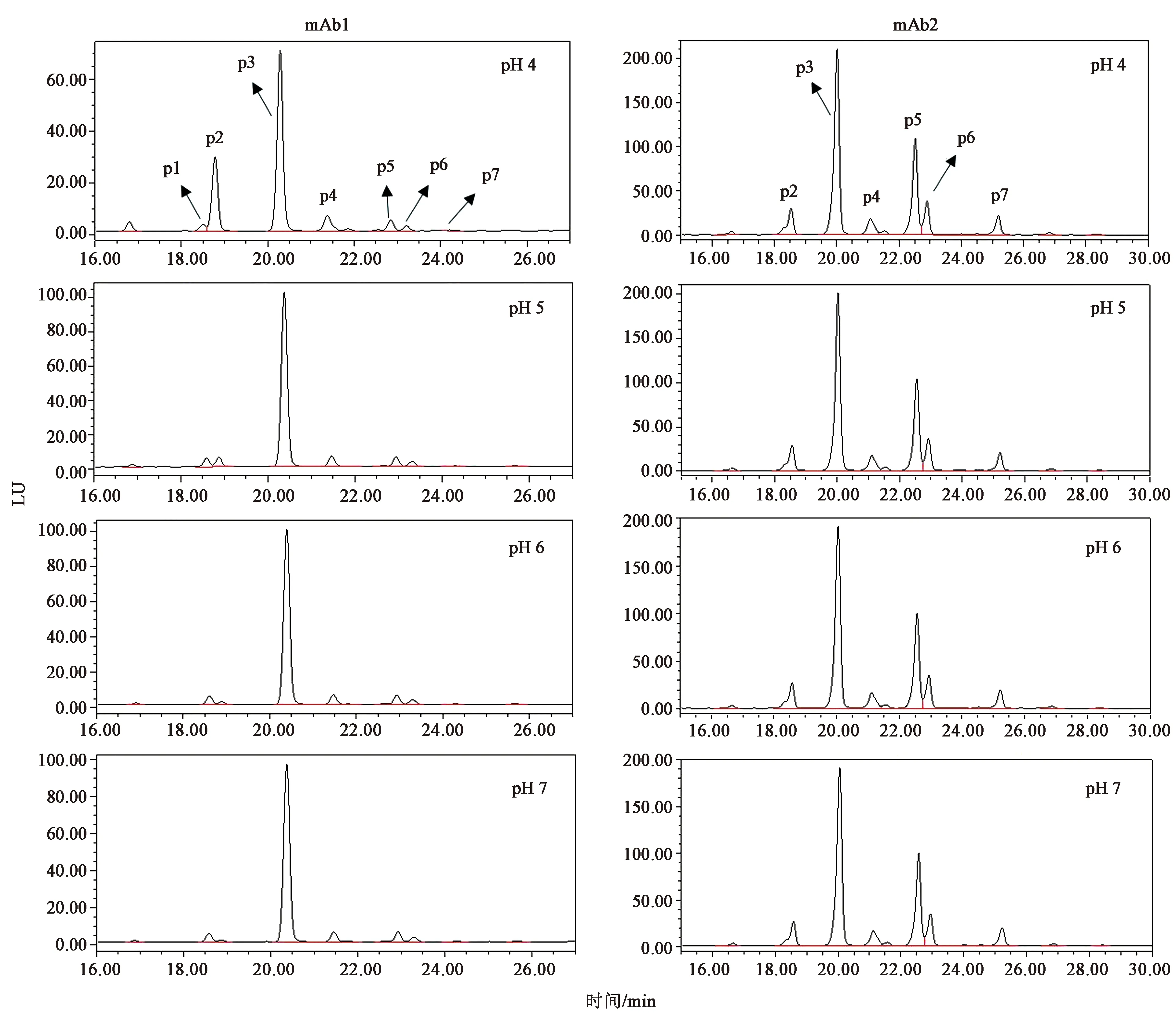
图1 mAb1和mAb2在不同pH缓冲液中酶解的N糖色谱图Fig.1 N-glycan chromatogram of mAb1 and mAb2 hydrolyzed in different pH buffer
将mAb1不同pH酶解后的蛋白进行还原CE-SDS检测,结果(图2)显示含N糖重链(HC)基本已被酶解成非糖基化重链(NG HC),说明不同pH缓冲液下PNGase F酶解N糖24 h后均酶解完全,N糖含量的变化并非不完全酶解造成。
利用液质联用鉴定各N糖的种类,p2和p3分别为G0F-GN(A1F)和G0F(A2F)。通过比较不同时间(1、8和24 h)酶解结果(表2),发现缓冲液pH为4和5时,p2和p3含量的变化随着酶解时间的增加而呈梯度变化,pH 4时尤为明显;pH为6和7时,变化不明显。因此推测在某些单抗分子上,G0F上的N-乙酰氨基葡萄糖在低pH条件下易从N糖链上脱落,使G0F形成G0F-GN,从而引起G0F-GN和G0F含量的变化。因此在进行PNGase F酶解时,应避免酶解体系中pH过低。
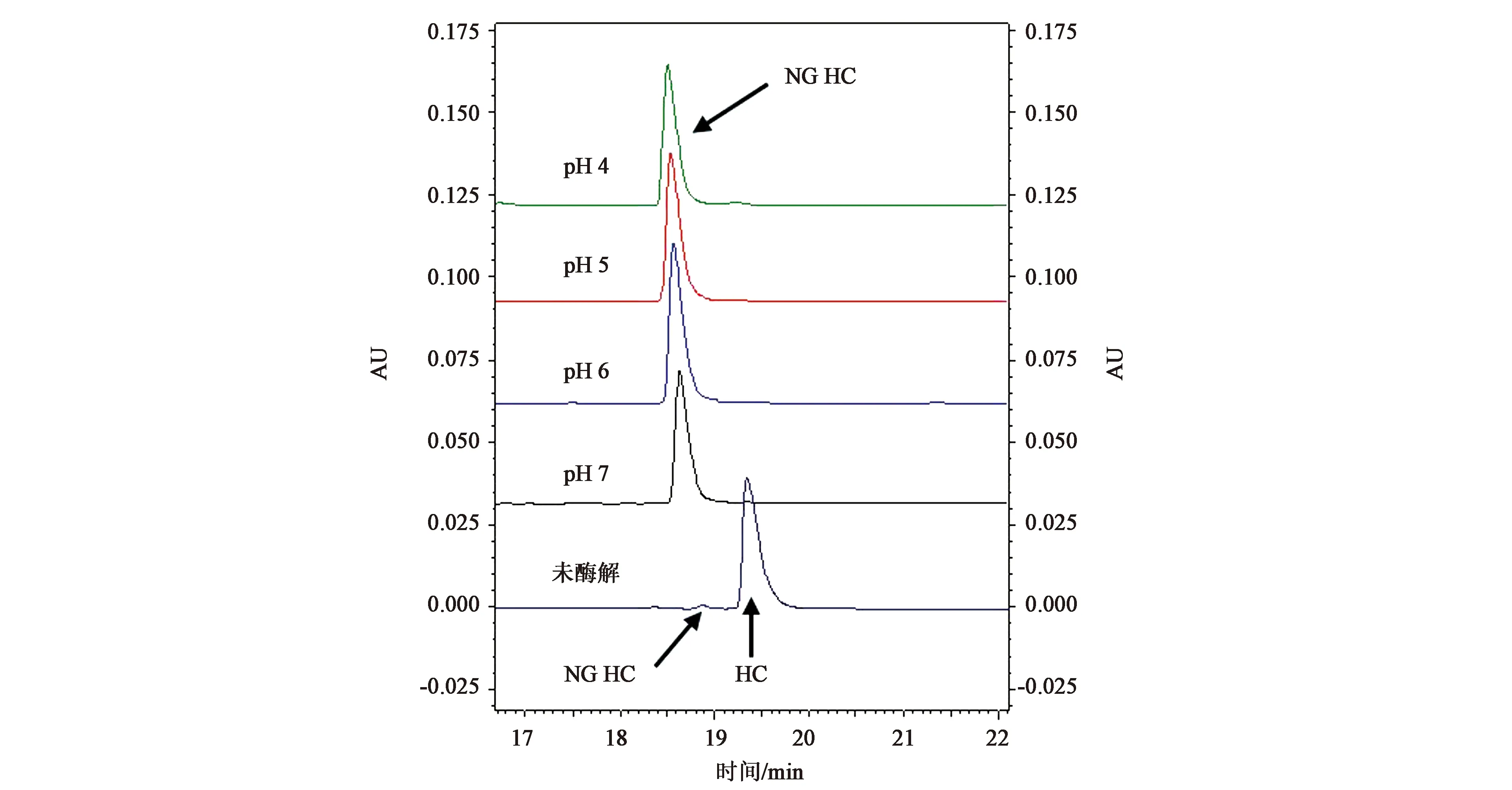
图2 mAb1不同pH酶解蛋白R CE-SDS电泳图Fig.2 Reduced CE-SDS of deglycoprotein from mAb1 hydrolyzed in different pH buffer
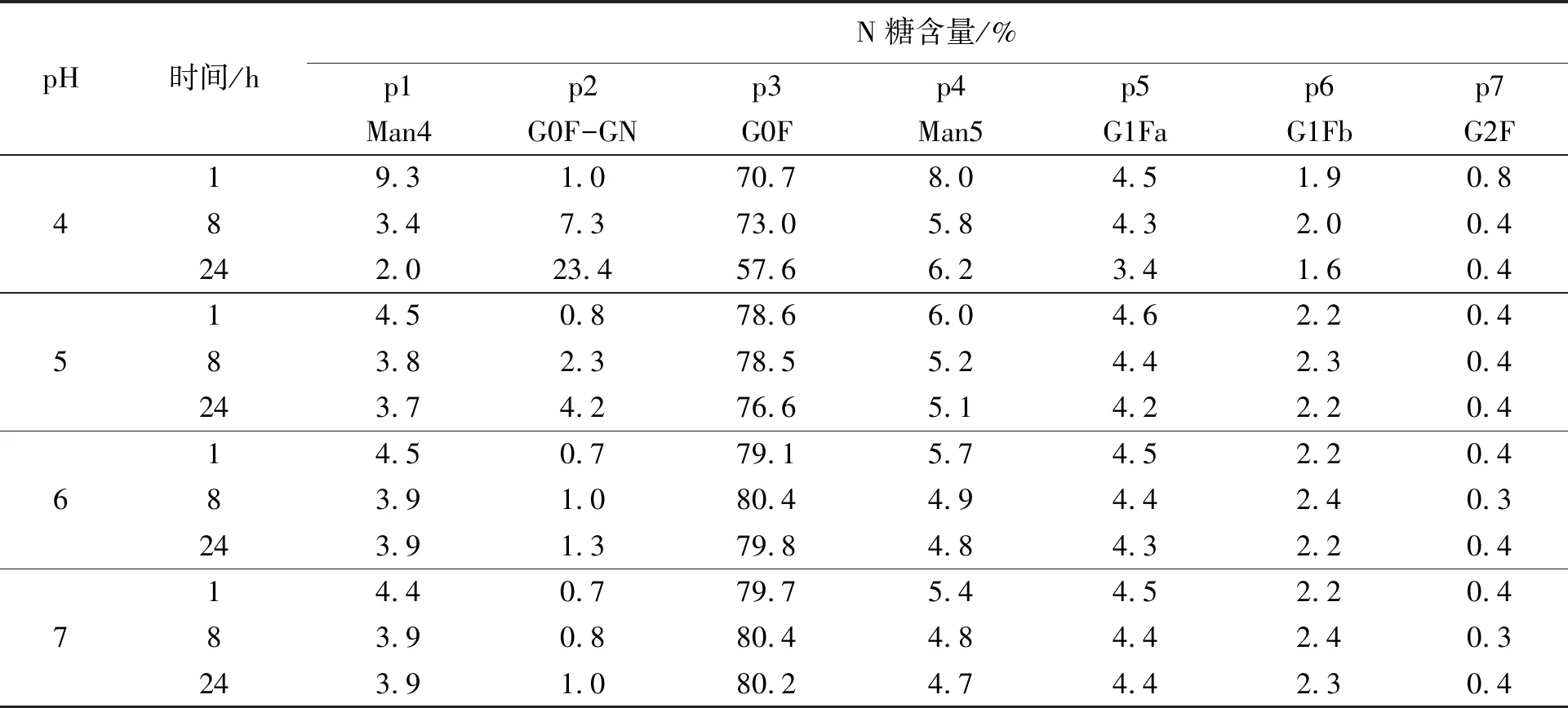
表2 mAb1在不同pH缓冲液中酶解不同时间的N糖含量Table 2 N-glycan content of mAb1 hydrolyzed in different pH buffer at different times
2.2 置换缓冲液的选择
为避免样品缓冲液pH及成分对PNGase F酶解的影响,用超滤离心管将样品换液至合适的溶液中再进行PNGase F酶解。本研究选择了1×PBS溶液(pH 7.4)和超纯水分别置换mAb1和mAb2的样品缓冲液,以未置换缓冲液(pH 7)为对照,PNGase F酶解24 h,进行N糖谱检测。N糖色谱图和含量分别见表3和图3,结果表明1×PBS溶液、超纯水和样品缓冲液(pH 7)的N糖谱结果一致。因为蛋白在超纯水中稳定性较差,所以本研究选用1×PBS溶液(pH 7.4)置换样品的缓冲液。
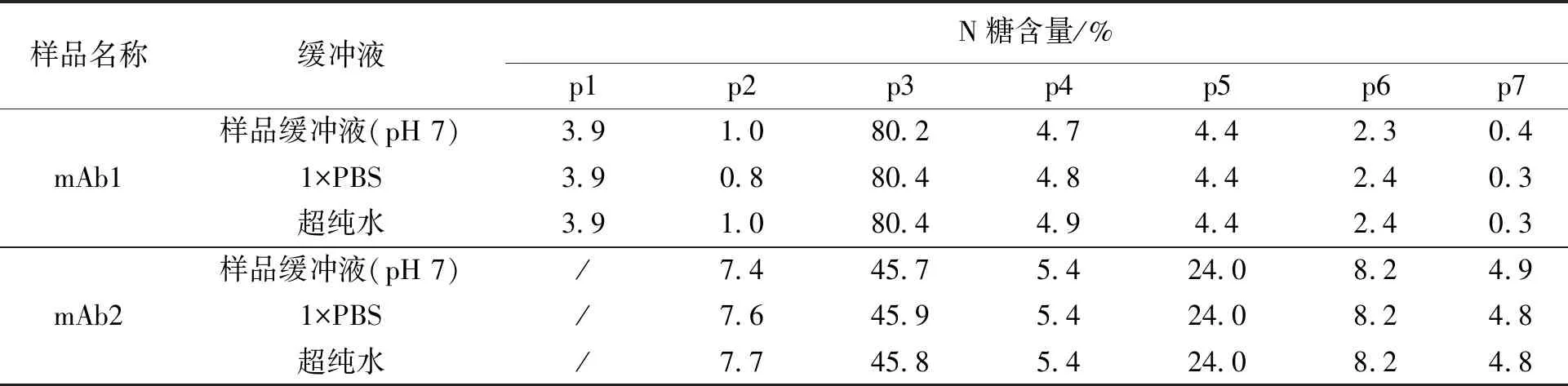
表3 mAb1和mAb2在样品缓冲液、1×PBS和超纯水中的N糖含量Table 3 N-glycan content of mAb1 and mAb2 hydrolyzed in sample buffer, 1×PBS and water

图3 mAb1和mAb2在样品缓冲液、1×PBS和超纯水中的N糖色谱图Fig.3 N-glycan profiles of mAb1 and mAb2 hydrolyzed in sample buffer, 1×PBS and water
2.3 不同PNGase F酶对N糖结果的影响
直接取200 μg样品,分别加入快速PNGase F酶(rapid PNGase F)和PNGase F进行酶解,N糖谱如图4所示。结果显示用rapid PNGase F进行酶解时,若选择用UPLC和1.7 μm粒径的ACQUITY UPLC Glycan BEH Amide Column色谱柱,则由于分离度的提高,mAb2的N糖色谱图(图4B)中G0F和G1Fa出现肩峰,而HPLC结果(图4A)由于分离度较差,此峰未得到有效分离。当将mAb2样品缓冲液置换至1×PBS中时(图4C),该肩峰消失。
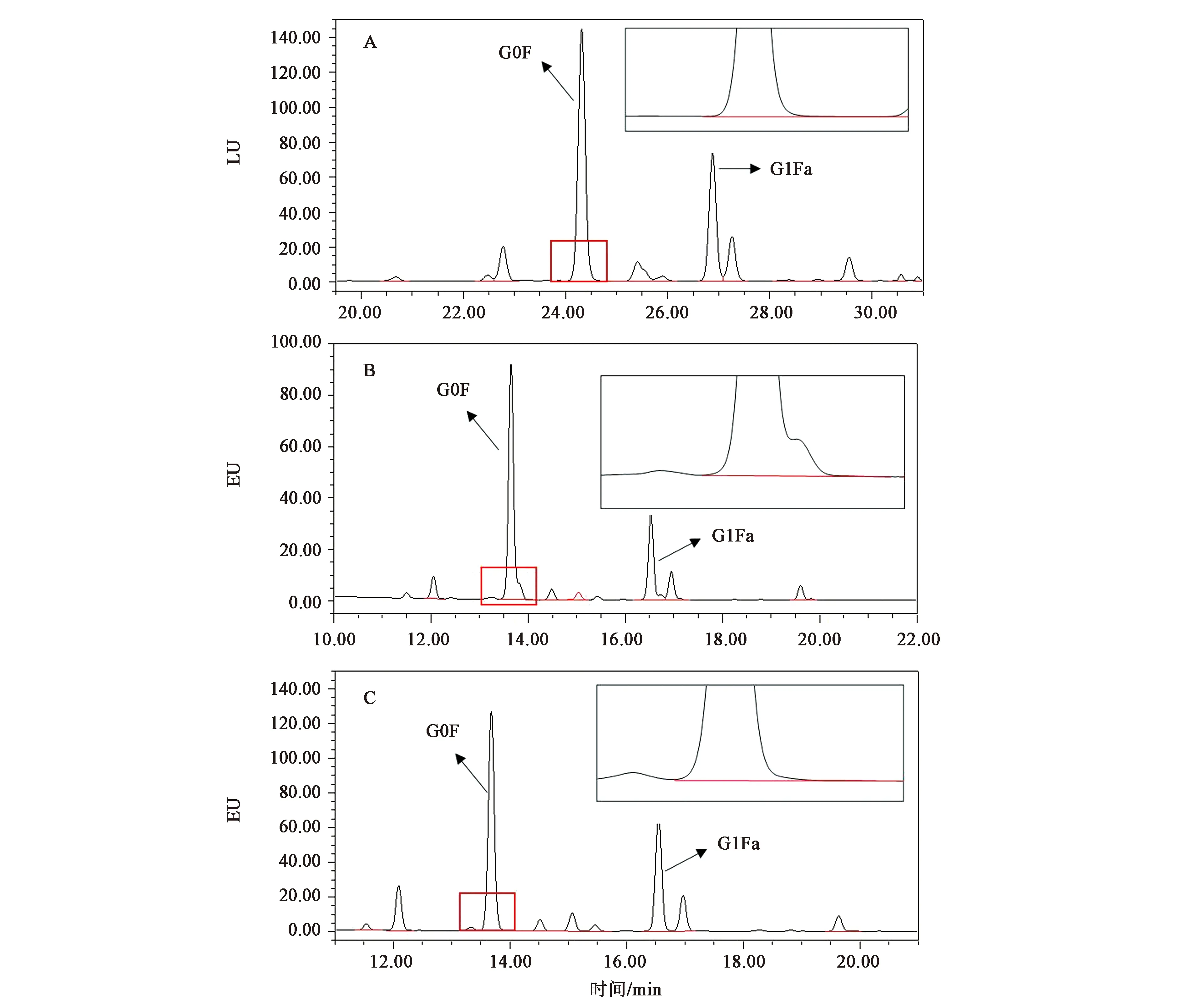
A:mAb2未置换缓冲液HPLC检测;B:mAb2未置换缓冲液UPLC检测;C:mAb2置换缓冲液UPLC检测。图4 不同PNGase F的N糖谱结果Fig.4 N-glycan profiles of mAb2 hydrolyzed by different PNGase F
综合2.1~2.3部分的研究结果可知,将样品置换缓冲液至合适的溶液中再进行PNGase F酶解,对改善N糖谱峰形和结果准确度至关重要,本研究采用了1×PBS溶液,效果良好。另外,在置换缓冲液的基础上使用快速PNGase F能将酶解时间缩短为数十分钟,有效提高了检测效率。选择UPLC能有效提高分辨率。
2.4 蛋白变性对酶解效率的影响
本研究发现,在非变性条件下利用快速PNGase F酶解不同单抗所需酶解时间差异明显,从数分钟至数小时不等。为了有效提高酶解效率,选择所需酶解时间最长的mAb2-F置换缓冲液至1×PBS中,加入变性缓冲液(5×)和1 μL rapid PNGase F酶,50℃孵育10 min后进行N糖谱检测处理,将沉淀的蛋白进行还原 CE-SDS检测,以未加变性缓冲液(5×)样品为对照。由图5还原CE-SDS结果可知,相同的酶解时间条件下,蛋白变性后由于高级结构被破坏,N糖充分暴露与酶接触,所以其酶解效果更好。
2.5 酶解程度对N糖结果的影响
在非变性条件下加入不同体积的rapid PNGase F酶对mAb2-F进行N糖谱检测处理,将沉淀的蛋白进行还原 CE-SDS检测,以加入变性缓冲液(5×)和1 μL rapid PNGase F酶的样品为对照,酶解条件均选择50℃、10 min。由图6(左)可知,在非变性条件下酶解相同时间,酶量的增加使非糖基化组分增多,但酶量增加至2 μL时酶解效果仍较差,而变性条件下用1 μL rapid PNGase F酶解,糖基化组分基本被转换成非糖基化组分。N糖水解的程度不同,N糖含量(表4)明显成梯度变化,说明PNGase F对不同类型的N糖水解效率不同,因此为了获得样品中真实的N糖含量水平,应将N糖尽量酶解完全。
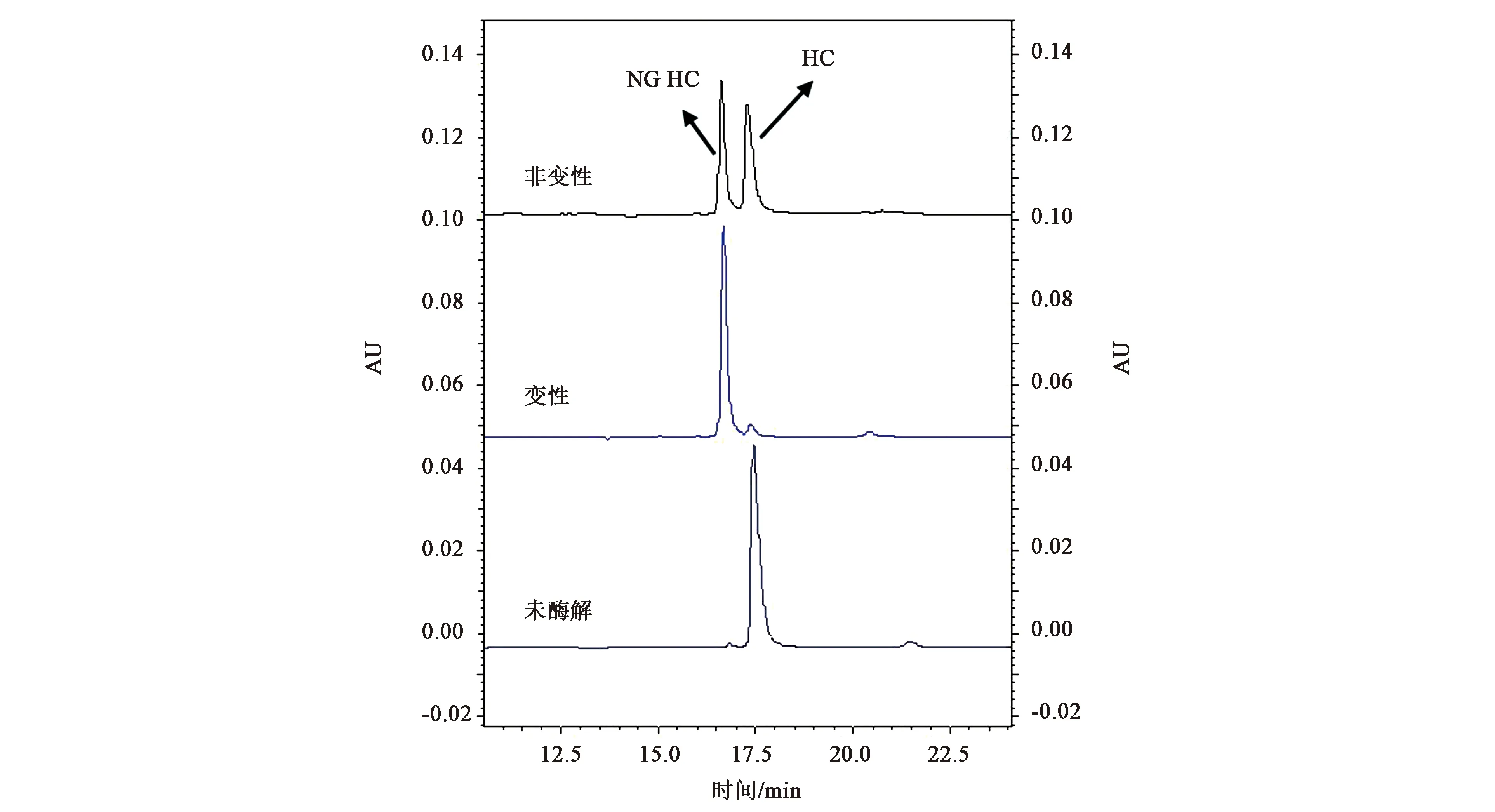
图5 mAb2-F在变性和非变性条件下切糖后还原CE-SDS图谱Fig.5 R CE-SDS profiles of mAb2-F deglycated in nature and denature conditions
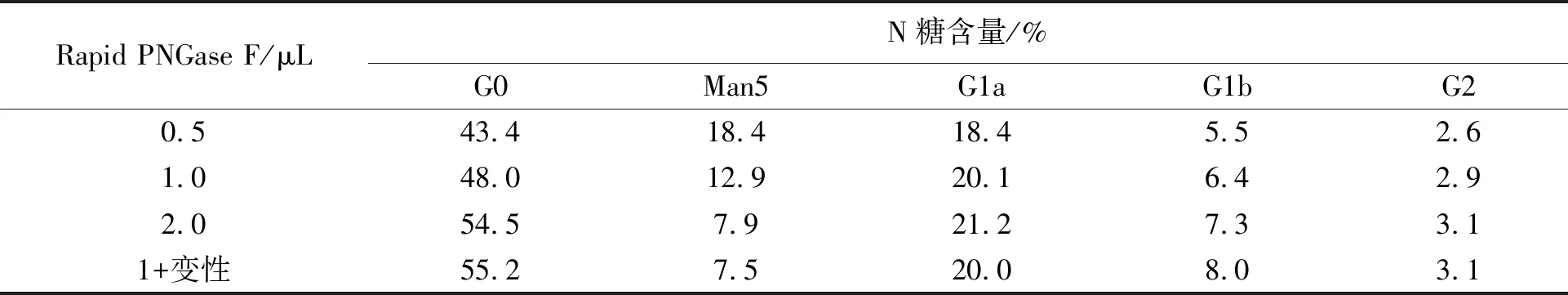
表4 不同体积rapid PNGase F酶解mAb2-F的N糖含量Table 4 N-glycan content of mAb2-F deglycated by different rapid PNGase F amount
3 讨论
N糖检测结果的准确性受多种因素的影响。在缓冲液pH对PNGase F酶解的影响研究中发现,低pH能使某些单抗在酶解过程中G0F逐渐转化为G0F-GN,造成检测结果的严重偏差,这种情况通常出现在Protein A亲和纯化后的样本,在低pH洗脱后直接进行酶解,若先将样品缓冲液pH调至中性或进行换液处理,能够避免pH的影响。对照mAb2置换和未置换样品缓冲液酶解结果,置换缓冲液能有效消除G0F和G1Fa的肩峰,消除样品缓冲液成分对检测结果的影响。
另外,本研究对多种单抗的研究发现,酶解程度不同的N糖检测结果存在明显差异,说明N糖苷酶F对不同类型的N糖水解效率不同,这与已有文献报道[20]一致。因此为了获得样品中真实的N糖含量水平,应将N糖尽量酶解完全。酶解程度可采用将蛋白沉淀进行还原CE-SDS检测,观察非糖基化组分含量的变化情况。
在细胞株筛选和工艺优化阶段,快速、准确的N糖检测结果能够有效推进开发进度,而该阶段巨大的样本量又对成本控制提出更高的要求,因此建立一种高效、准确且低成本的N糖检测方法尤为重要。本研究确定了较为通用的PNGase F酶对单克隆抗体的酶解条件,即为将样品置换缓冲液至1×PBS,取适量样品加入变性缓冲液(5×)和1 μL Rapid PNGase F酶,50℃孵育10 min,进行后续除蛋白、干燥等处理,快速PNGase F酶和变性处理的应用,将酶解时间大幅缩短,提高了检测效率;同时采用传统的UPLC和ACQUITY UPLC Glycan BEH Amide Column色谱柱,有效提高了分离度,并控制了检测成本。
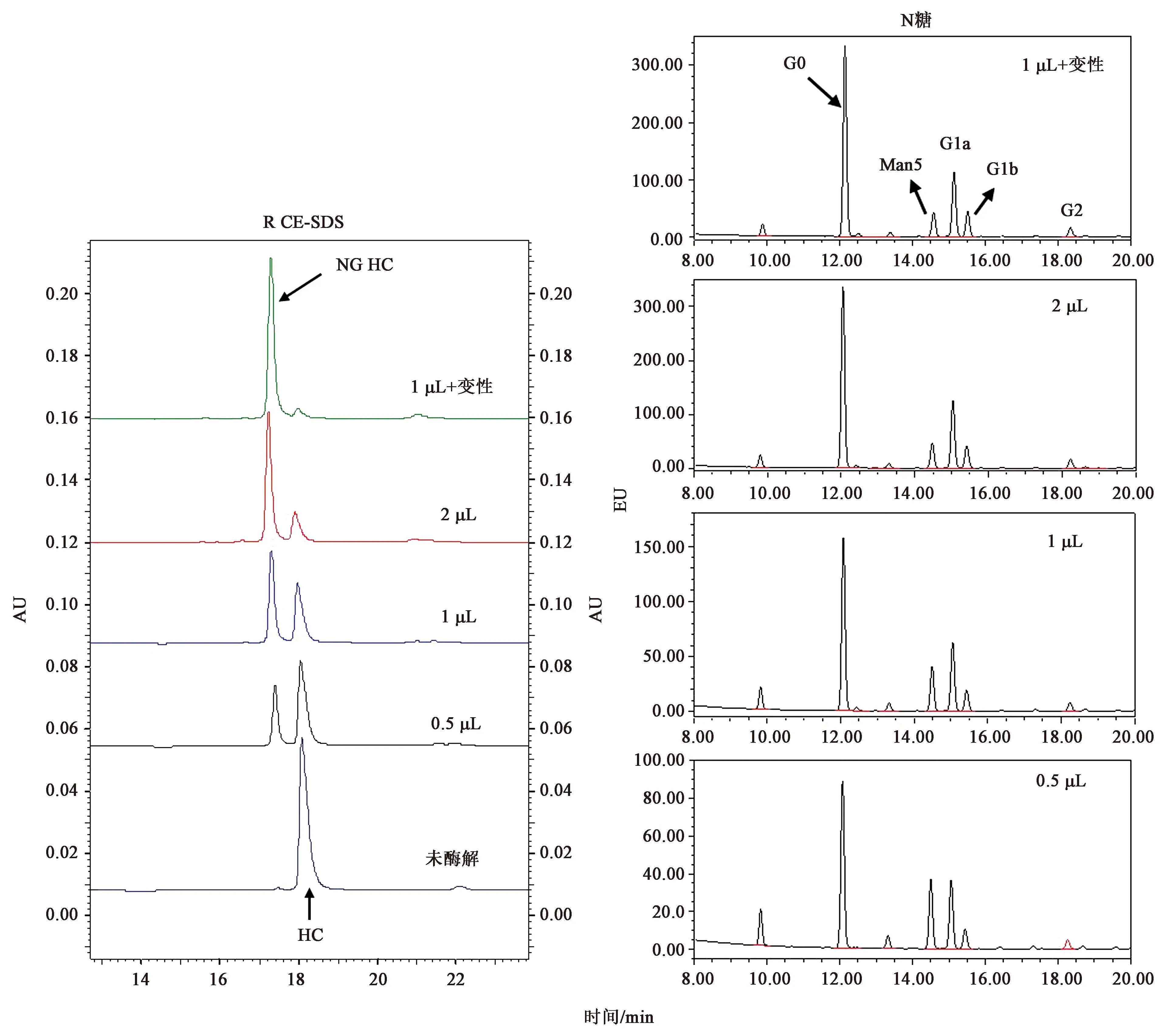
图6 不同体积rapid PNGase F酶解mAb2-F的R CE-SDS和N糖谱图Fig.6 N-glycan and R CE-SDS profiles of mAb2-F deglycated by different rapid PNGase F amount
猜你喜欢
中国造纸(2022年9期)2022-11-25
中国造纸(2022年8期)2022-11-24
复旦学报(医学版)(2021年5期)2021-10-13
中国科技纵横(2021年24期)2021-03-02
蚕桑通报(2020年1期)2020-07-10
环球时报(2019-04-03)2019-04-03
癌症进展(2018年11期)2018-12-30
中国土壤与肥料(2018年5期)2018-11-05
中成药(2018年6期)2018-07-11
医学研究杂志(2015年7期)2015-06-22
