
NLRP3炎症小体激活机制及其在脓毒症中的作用
2021-03-27 01:50吴俊东耿智隆杨勇丽杨婉君张强英
医学综述 2021年3期
吴俊东,耿智隆,杨勇丽,杨婉君,张强英
(1.兰州大学第二临床医学院麻醉学系,兰州730030; 2.联勤保障部队第九四〇医院麻醉科,兰州 730050)
脓毒症是由感染引起的宿主反应失调导致的危及生命的器官功能障碍综合征[1]。有文献报道,我国外科重症监护病房及综合重症监护病房严重脓毒症患病率分别为8.7%和37.3%;虽然不同国家和地区的发病率有所不同,但脓毒症总体发病率仍有逐年上升趋势,随着医疗水平不断提高,脓毒症的预后已有极大改善,但病死率仍为30%~50%[2]。因此,明确脓毒症的发病机制,对防治脓毒症尤为关键。机体炎症反应失控是脓毒症发病的重要机制,炎症小体在其中的作用受到越来越多的关注。炎症小体是一种由核苷酸结合寡聚化结构域样受体家族成员与PYHIN (pyrin and HIN domain)家族成员组成的胞质多蛋白复合物,其中核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)备受关注。NLRP3炎症小体参与多种疾病的炎症、免疫和代谢进程,是一种重要的先天免疫传感器,可以对结构多样的损伤相关分子模式做出反应,诱发炎症反应[3]。NLRP3炎症小体激活在脓毒症及其多器官功能障碍中扮演重要角色。现就NLRP3炎症小体的激活机制及其在脓毒症中的作用予以综述。
1 NLRP3炎症小体的激活机制
1.1离子调节 离子调节包括K+外流、Na+内流、Cl-外流和Ca2+信号转导,已被确定为NLRP3炎症小体激活的关键因素[4-6]。其中,K+浓度降低是NLRP3炎症小体活化的触发因素,已知有许多NLRP3炎症小体的激活因子(包括成孔毒素、尼日利亚菌素、腺苷三磷酸和颗粒物质)可诱导K+外流,由此认为细胞内K+减少是NLRP3炎症小体激活的共同机制[4]。有研究指出,NIMA相关蛋白激酶7(NIMA-related kinase 7,NEK7)可以直接与NLRP3蛋白结合,在K+外流的下游发挥调节NLRP3炎症小体组装和激活的作用[7]。研究表明,双孔结构域K+通道TWIK2是触发NLRP3炎症小体激活的K+外流通道[8],使人们对K+外流激活NLRP3有了更进一步的认识。但有研究表明,咪喹莫特以及相关分子CL097可诱导活性氧类(reactive oxygen species,ROS)产生,促进NLRP3炎症小体活化,而不依赖于K+外流[9]。因此,K+外流在NLRP3炎症小体激活中是一个重要但非必需的因素。
据报道,Ca2+信号通路是NLRP3炎症小体活化的重要组成部分,抑制Ca2+动员可降低NLRP3炎症小体的活化[5]。同时,Lee等[10]指出,通过钙敏感受体触发磷脂酶C产生1,4,5-三磷酸肌醇,而1,4,5-三磷酸肌醇与其受体相互作用,促进内质网释放Ca2+,从而激活NLRP3炎症小体。还有研究显示,钙敏感受体相关家族成员6A也具有相似作用[11]。细胞内Ca2+信号通路通过触发线粒体损伤或细胞内释放的Ca2+通过钙调蛋白依赖性蛋白激酶Ⅱ激活转化生长因子-β激活激酶1/c-Jun氨基端激酶通路,调控NLRP3炎症小体的激活[12]。但另有研究通过对比K+外流和Ca2+内流的作用发现,Ca2+信号通路在NLRP3炎症小体激活中不是必需的[4]。有研究表明,K+外流和Ca2+内流对线粒体ROS的生成至关重要,但K+外流是Ca2+内流的必要条件,从而触发NLRP3炎症小体[13]。因此,需要进一步研究Ca2+信号在NLRP3炎症小体活化中的作用机制。
在炎症通路中Cl-的作用也常被关注。有研究表明,非甾体抗炎药通过抑制容量调节阴离子通道的Cl-外流来阻止NLRP3炎症小体激活[14]。Tang等[6]报道了细胞内Cl-通道(chloride intracellular channel,CLIC)在NLRP3炎症小体调控中的作用,CLIC作用于K+外流-线粒体ROS轴下游,可促进NLRP3炎症小体的激活,而NLRP3激动剂可诱导K+外流,导致线粒体损伤和ROS生成;线粒体ROS可诱导CLICs转位至质膜,诱导Cl-外流,促进NEK7-NLRP3相互作用、炎症小体组装、胱天蛋白酶(caspase)-1激活和白细胞介素(interleukin,IL)-1β分泌。Green等[15]的进一步研究证明了Cl-外流是凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a caspase activation and recruitment domain,ASC)的寡聚信号,激活caspase-1和分泌IL-1β需要K+外流依赖的NEK7激活与NLRP3的相互作用。有报道显示,CLIC1和CLIC4也参与NLRP3炎症小体的调控,CLIC1或CLIC4小干扰RNA转染抑制了IL-1β转录、ASC斑点形成以及成熟IL-1β的分泌[16]。容量调节阴离子通道与CLIC之间是否也存在某种联系值得进一步探讨。有报道指出,Na+内流参与了NLRP3炎症小体活化,但Na+内流和细胞内K+外流联动才能激活NLRP3炎症小体,且莫能菌素诱导的单纯Na+内流并未导致NLRP3炎症小体的激活[4]。因此,Na+内流可能不是NLRP3炎症小体激活所必需的。
1.2溶酶体 Hornung等[17]证明,摄取内源性或外源性晶体物质(如二氧化硅和明矾),可诱导溶酶体肿胀、渗漏和组织蛋白酶B释放,触发NLRP3炎症小体激活。Weber和Schilling[18]发现,在NLRP3炎症小体活化过程中,溶酶体钙信号通过IL-1β信使RNA稳定机制调控IL-1β前体的产生(启动步骤),而溶酶体组织蛋白酶B则介导NLRP3-caspase-1 复合物的激活(组装步骤)。Okada等[12]研究发现,溶酶体破裂激活的Ca2+-钙调蛋白依赖性蛋白激酶Ⅱ激活转化生长因子-β激活激酶1/c-Jun氨基端激酶途径通过促进ASC寡聚化,调节NLRP3炎症小体的激活,表明溶酶体破裂和Ca2+流通共同促进NLRP3的活化。Muoz-Planillo等[4]报道,颗粒物通过吞噬作用导致溶酶体膜损伤,且这种损伤通过打开一个或多个可渗透K+的膜孔,触发K+外溢引起NLRP3炎症小体活化;同时还发现,脂多糖(lipopolysaccharide,LPS)诱导可能会增强由晶体或溶酶体抑制剂引起的K+外流。由此可见,溶酶体和离子流通在激活NLRP3炎症小体方面可能存在协同作用。
1.3ROS生成 ROS生成是NLRP3炎症小体激活的重要因素,但目前还存在诸多争议。许多物质可诱导不同类型细胞线粒体ROS生成,如介孔二氧化硅纳米粒子可诱导肝细胞产生ROS,从而激活NLRP3,ROS清除剂则可以显著改善肝细胞中NLRP3的活化[19]。有研究指出,咪喹莫特诱导ROS促进NLRP3炎症小体活化,但如果细胞缺乏NEK7,咪喹莫特的刺激则不能产生IL-1β[9]。Tschopp和Schroder[20]指出,ROS的积累能够释放对ROS敏感的NLRP3配体,即硫氧还蛋白相互作用蛋白,导致NLRP3炎症小体活化,但在没有硫氧还蛋白相互作用蛋白的情况下,caspase-1的激活并未完全被阻断。Li等[21]报道,线粒体ROS和NLRP3炎症小体均参与臭氧诱导的肺部炎症反应,但只有NLRP3参与臭氧诱导的肺气肿,说明NLRP3炎症小体可能被非线粒体ROS机制激活。因此,需要进一步研究阐明ROS在NLRP3炎症小体活化中的确切作用。
1.4NLRP3翻译后修饰 NLRP3翻译后修饰包括磷酸化和泛素化,在NLRP3炎症小体活化中起重要作用。Song等[22]研究显示,JNK1介导的NLRP3磷酸化是一个关键的分子启动事件,阻断其S194位点磷酸化可阻止Cryopyrin蛋白相关周期综合征中NLRP3的激活。Stutz等[23]证明,NLRP3的激活由其蛋白结构域的磷酸化控制,且激酶和磷酸酶的平衡在其中起重要作用。除丝氨酸外,酪氨酸磷酸化同样参与了NLRP3的调控。Spalinger等[24]研究表明,蛋白酪氨酸磷酸酶非受体型22与NLRP3相互作用,使蛋白酪氨酸磷酸酶非受体型22在Tyr861位点去磷酸化,从而促使NLRP3炎症小体激活和IL-1β分泌。
蛋白质的磷酸化不仅可以直接影响NLRP3的活化,还可以通过作用于泛素化间接调控NLRP3的激活。研究表明,胆汁酸通过G蛋白偶联胆汁酸受体5信号通路磷酸化NLRP3蛋白第291位丝氨酸,并增强NLRP3蛋白泛素化水平,最终抑制NLRP3炎症小体激活[25]。Yan等[26]研究显示,神经递质多巴胺通过多巴胺D1受体抑制NLRP3炎症小体的激活,多巴胺D1受体信号通过第二信使环腺苷酸负调控NLRP3炎症小体,环腺苷酸与NLRP3结合,并通过E3泛素化连接酶MARCH7(membrane-associated RING-CH-type finger 7)促进多巴胺D1受体泛素化和降解。E3泛素连接酶TRIM31(tripartite motif containing 31)和ARIH2(ariadne homolog 2)也被相继报道在NLRP3通路中有重要作用[27-28]。一项研究指出,ABR01(abraxas brother 1)是BRISC(BRCC36 isopeptidase complex)去泛素酶复合物的一个亚基,高效的NLRP3活化需要ABRO1的参与,LPS启动诱导ABRO1以S194磷酸化依赖的方式与NLRP3结合,随后在激活剂的刺激下招募BRISC移除NLRP3的K63连接的泛素链[29]。因此,NLRP3的磷酸化和泛素化在NLRP3炎症小体活化调控中起关键作用,但具体机制仍需更多的研究。研究其他翻译后修饰(如甲基化和乙酰化)在NLRP3炎症小体活化调控中的作用可能是一个新的切入点。
除上述调节机制外,还存在非经典途径炎症小体激活,胆碱的吸收和代谢过程也可促进NLRP3炎症小体的活化。总之,NLRP3炎症小体的激活是一个复杂的过程,只有明确其作用机制,才能针对性地开发药物和使用药物,防治NLRP3炎症小体通路作用的相关疾病。NLRP3炎症小体激活机制见图1。
2 NLRP3炎症小体与脓毒症
在脓毒症发生的最初阶段炎症因子风暴发挥重要作用。IL-1β等细胞因子的合成和分泌是诱导全身炎症反应的关键因素,同时也是机体清除病原微生物的重要过程。因此,适当的炎症反应对机体是有利的,但过度的炎症反应会造成细胞和组织损伤,引发脓毒症甚至脓毒性休克。IL-1β的成熟和活化受caspase-1调控,而caspase-1的调控与NLRP3炎症小体密切相关。
研究报道,脓毒症时NLRP3炎症小体的激活可引起组织线粒体功能障碍并诱发炎症反应,而自噬介导的NLRP3炎症小体失活可减轻脓毒症引起的炎症反应和器官损伤[3]。Lee等[30]指出,NLRP3基因缺陷可有效抑制盲肠结扎穿孔(cecal ligation and puncture,CLP)脓毒症模型小鼠的炎症反应,提高其存活率;可能的机制为NLRP3基因缺陷减轻了caspase-7对脂氧素B4生物合成的抑制,并通过脂氧素B4的产生抑制促炎细胞因子,从而促进多菌败血症炎症的消除,提高存活率。Rahim等[31]研究发现,褪黑激素治疗和剔除NLRP3基因对CLP模型小鼠具有相似的作用,均能抑制NLRP3炎症小体的激活,进而减轻炎症反应。另外,炎症小体的活化产物同样也不容忽视,NLRP3炎症小体的激活调控caspase-1的活化,从而促进IL-1β和IL-18的产生,而IL-18对感染性休克病死率的预测可能有重要作用[32]。有实验证实,在剔除caspase-1的情况下,CLP模型小鼠假单胞菌的清除并未发生障碍,但CLP损伤后caspase-1表达升高和活性增加可能导致先天免疫功能降低[33],这使得脓毒症免疫紊乱发病机制更具说服力。同时,Meissner等[34]指出,超氧化物歧化酶1能够调控caspase-1的激活,在体内实验过程中发现,超氧化物歧化酶1缺乏的小鼠产生较少的caspase-1依赖性细胞因子,对LPS诱导的感染性休克也不敏感。然而,Vanden Berghe等[35]认为,在脓毒症中同时靶向IL-1和IL-18才能有效提高生存率,而不是仅仅抑制上游的caspase-1,这为靶向上游NLRP3炎症小体提供了新思路。综上,靶向调控NLRP3炎症小体通路对脓毒症的预防、治疗和转归可能具有积极的作用。
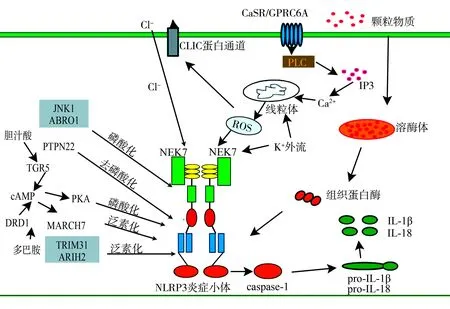
NLRP3:核苷酸结合寡聚化结构域样受体蛋白3;CaSR:钙敏感受体;GPRC6A:CaSR相关家族成员6A;CLIC:细胞内氯离子通道;IP3:三磷酸肌醇;ROS:活性氧类;JNK1:c-Jun氨基端激酶1;ABRO1:abraxas brother 1;PTPN22:蛋白酪氨酸磷酸酶非受体型22;NEK7:NIMA相关蛋白激酶7;TGR5:G蛋白偶联胆汁酸受体5;cAMP:环腺苷酸;PKA:蛋白激酶A;DRD1:多巴胺D1受体;MARCH7:membrane-associated RING-CH-type finger 7;TRIM31:tripartite motif containing 31;ARIH2:ariadne homolog 2;caspase-1:胱天蛋白酶1;IL:白细胞介素
3 NLRP3与脓毒症后多器官功能障碍
2016年国际专家组将脓毒症定义为感染引起的宿主反应失调所致的危及生命的器官功能障碍,而不单纯是全身炎症反应,同时强调器官功能障碍[1]。器官功能障碍是脓毒症的严重并发症,是患者预后不良的主要原因,发病机制复杂,炎症反应可能是其发病机制之一。NLRP3炎症小体在炎症反应中扮演重要角色。
3.1NLRP3与脓毒症心肌病 脓毒症性心肌病是脓毒症的严重并发症,与重症监护病房的高病死率有关。因此,治疗脓毒症心肌病对改善脓毒症患者的预后至关重要。但脓毒症性心肌病的具体病因目前尚不明确,可能与过度的炎症反应有关。研究显示,脓毒症时心脏成纤维细胞中的NLRP3炎症小体被激活,诱导IL-1β等炎症因子成熟和释放,通过抑制心肌成纤维细胞NLRP3炎症小体激活可以减轻LPS诱导的小鼠心肌功能障碍,提高腹膜炎型脓毒症小鼠的存活率[36]。同时,一氧化碳释放分子通过阻断NLRP3炎症小体与适应性蛋白ASC的相互作用,抑制NLRP3炎症小体的活化,减轻脓毒症小鼠的心肌功能障碍[37]。此外,Luo等[38]研究表明,通过阻断NLRP3通路,可以减轻LPS诱导的炎症反应和细胞凋亡,恢复受损的心肌功能。心脏是循环系统的动力器官,心肌功能障碍会影响机体循环,破坏全身血流动力学稳定。因此,探究NLRP3炎症小体在脓毒症心肌功能障碍中的作用,可能为逆转心肌功能、恢复全身组织血流灌注、开发新的治疗靶点提供依据。
3.2NLRP3与脓毒症肺损伤 脓毒症患者的肺组织往往是最先受累的器官,进而诱发急性肺损伤(acute lung injury,ALI)和急性呼吸窘迫综合征。有证据表明,NLRP3炎症小体活化在ALI中发挥重要作用[39]。研究显示,在LPS诱导的ALI中,NLRP3炎症小体可促进caspase-1激活和IL-1β表达,其可能机制为褪黑素通过抑制细胞外组蛋白的释放或直接阻断组蛋白诱导NLRP3炎症小体激活[40]。在脓毒症诱发的急性呼吸窘迫综合征中发现,支气管上皮细胞NLRP3炎症小体通路激活导致支气管上皮细胞分泌过量黏蛋白和炎症细胞因子,促进趋化因子和细胞黏附分子的表达[41]。Wang等[42]报道,二氢杨梅素可显著抑制CLP脓毒症模型小鼠的NLRP3炎症小体活化,并抑制肺内细胞焦亡相关蛋白的表达,从而保护CLP诱导的ALI。Luo等[43]指出,氯化血红素可抑制NLRP3炎症小体的活化,主要机制是氯化血红素能够增加血红素加氧酶-1蛋白的表达和活性,减少IL-1β和IL-18的分泌,减轻炎症反应。但该研究并未指明血红素加氧酶-1与NLRP3相互作用的机制,还有待进一步研究。综上可见,通过抑制NLRP3炎症小体的活化能有效缓解ALI,但如何直接阻断ALI中NLRP3炎症小体的激活目前仍不清楚。
3.3NLRP3与脓毒症肾损伤 在脓毒症,尤其是严重脓毒症或脓毒性休克中,极易伴发急性肾损伤(acute kidney injury,AKI)。有报道称,在最初的脓毒症、缺血或肾毒性损伤后,肾内皮细胞和肾小管上皮细胞释放炎症细胞因子和趋化因子,导致白细胞募集和肾损伤放大,炎症小体在这一过程中发挥重要作用[44]。研究显示,在LPS诱导的脓毒症模型小鼠中NLRP3、caspase-1的表达均显著升高,而通过金丝桃素的处理可有效降低脓毒症小鼠血清尿素氮和肌酐水平,进而保护肾功能,其机制可能是通过抑制Toll样受体4和NLRP3信号通路,减少肿瘤坏死因子-α和IL-1β等炎症因子的表达[45]。有研究指出,外源性一氧化碳能够抑制NLRP3炎症小体活性,显著降低CLP小鼠模型中肿瘤坏死因子-α和IL-1β水平,从而预防脓毒症引起的AKI[46]。Zhao等[47]研究表明,在CLP小鼠模型中,沉默信息调节因子3通过降低ROS的产生,抑制NRLP3炎症小体,减轻氧化应激,下调IL-1β和IL-18表达,对肾脏线粒体损伤起保护作用。由此可知,炎症反应、氧化应激和线粒体损伤均参与了脓毒症引发的AKI,若能全面了解氧化应激、线粒体损伤与Toll样受体4及NRLP3信号通路在AKI中的具体作用,联合运用多靶点治疗,可能为临床治疗脓毒症及AKI提供新的可能。
综上所述,在脓毒症的多器官损伤中NRLP3炎症小体信号通路扮演重要角色,但目前NRLP3炎症小体在脓毒症肝、脑、肠等器官损伤中的研究较少。因此,NRLP3炎症小体在器官损伤中的作用值得进一步发掘。脓毒症发病机制复杂,仅通过阻断炎症小体通路预防脓毒症多器官损伤是否可靠仍需要更深入的研究。
4 小 结
炎症小体在先天性免疫及免疫炎症性疾病的发生、发展中发挥关键作用。近年来NLRP3炎症小体成为研究热点,但关于NLRP3炎症小体组装及活化的机制仍有待进一步研究。研究人员针对NLRP3/ASC/caspase-1/IL-1β/IL-18炎症通路研发的一些特异性抑制剂,为脓毒症和其他炎症性疾病的治疗提供了新的可能。但目前大多研究为动物实验,未来能否在人类受益仍需要大样本、多中心、前瞻性的临床试验。总之,对NLRP3炎症小体在脓毒症中作用研究的不断深入,可能为脓毒症的发病机制及治疗提供新的思路。
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国临床解剖学杂志(2022年1期)2022-11-15
中国中医急症(2022年9期)2022-10-12
重庆医学(2022年14期)2022-08-06
检验医学与临床(2022年12期)2022-06-27
现代检验医学杂志(2022年3期)2022-06-27
汽车实用技术(2022年10期)2022-06-09
现代临床医学(2022年2期)2022-04-19
滨州医学院学报(2021年2期)2021-05-13
领导文萃(2018年13期)2018-08-13
