
高效液相色谱法同时测定牙膏及漱口水中的23种致癌染料
2021-03-25 02:48费晓庆丁友超董绍伟曹丽华
分析测试学报 2021年3期
汤 娟,费晓庆,富 薇,周 佳,丁友超,钱 凯,董绍伟,曹丽华
(1.南京海关工业产品检测中心,江苏 南京 210019;2.南京海关动植物与食品检测中心,江苏 南京 210019;3.中国合格评定国家认可中心,北京 100062;4.南京金检检验有限公司,江苏 南京 210019)
根据心理学研究分析,人类通过感觉接受的外界信息主要来自于视觉,其中颜色尤其是鲜艳的颜色尤为重要,因此在牙膏和漱口水的生产过程中,生产者会通过加入染料来吸引更多消费者。牙膏中常见的染料有二氧化钛(Cl 77891)、酸性红33(Cl 17200)、柠檬黄(Cl 19140)、亮蓝(Cl 42090)、颜料绿7(Cl 74260)等,漱口水中常见的染料有亮蓝、柠檬黄、胭脂红等。染料可分为天然染料和合成染料,目前已知的染料有七千多种,牙膏和漱口水中常见染料以合成染料为主。然而,随着合成染料的大量使用和科学技术的发展,研究发现一些合成染料对人、动物、植物和环境会造成不可逆的损伤,如:酸性红26、直接红28和分散蓝1等染料含有芳香胺结构,具有直接致癌诱变性;碱性红9与男性膀胱癌的发生有关联,对哺乳动物还可诱发肝癌、甲状腺癌、乳腺癌和淋巴癌等[1]。我国《化妆品监督管理条例》[2]规定将牙膏参照普通化妆品的相关要求进行管理,《化妆品安全技术规范》[3]中明确将分散蓝1、分散黄3、碱性红9、碱性紫3、直接黑38、直接蓝6、直接棕95、直接红28、溶剂黄1、溶剂黄3、酸性紫49、分散橙149等致癌染料列为禁用染料,但该技术法规未包含所有禁用致癌染料。
此外,世界卫生组织国际癌症研究机构(IARC)将酸性红26、酸性红114、直接蓝15、碱性紫14等染料列入2B类致癌物清单[4]中;欧盟化学品管理局(ECHA)公布的高关注度物质(SVHC)清单[5]中将碱性蓝26等染料的属性定为致癌;ISO 16373-2:2014[6]将分散橙11、溶剂黄2等归类为致癌染料;服装及鞋袜国际物质限用清单管理工作组(AFIRM-RSL)[7]将分散黄23、分散黄56、分散黄7、分散红151列为禁用染料。综上,建立灵敏、有效的测定牙膏和漱口水中多种致癌染料的分析方法具有重要意义和实用价值。
目前针对致癌染料检测的基质集中于纺织品[8-9]、皮革[10]、染料产品[11-12]、塑料制品[13]和玩具[14-15]等,检测方法主要有液相色谱法(LC)[16-17]和液相色谱-质谱法(LC-MS)[18-20],但与牙膏和漱口水相关的研究较为缺乏。LC-MS法与LC法相比虽具有更高的灵敏度,但牙膏中的表面活性剂和增稠剂会对质谱造成严重污染。基于前文提及的各种法规和清单,本文采用高效液相色谱法对牙膏和漱口水中的23种致癌染料进行测定,目标化合物个数远高于文献报道。本方法可为牙膏和漱口水的品质安全监管提供技术支持,具有较高的应用价值。
1 实验部分
1.1 仪器、试剂与材料
1260型高效液相色谱仪,配备二极管阵列检测器(DAD,美国Agilent公司);KQ-250DB型数控超声波清洗器(昆山市超声仪器有限公司);Milli-Q去离子发生器(美国Sigma公司);T215/IPC 250-3真空干燥器(上海领德仪器有限公司);XW-80A微型涡旋混合仪(上海沪西分析仪器厂有限公司);PL602-L和ML54型电子天平(感量0.01 g和0.000 1 g,梅特勒-托利多上海有限公司)。
甲醇、乙腈和乙醇(色谱纯,德国Merck公司);色谱纯乙酸铵、0.5 mol/L磷酸二氢四丁基铵(美国Sigma-Aldrich公司);70%~75%乙酸二正己基铵、0.5 mol/L四丁基氟化铵(日本TCI公司);石英砂(分析纯,南京化学试剂股份有限公司)。
标准品:碱性红9(78.3%,纯度,下同)、碱性紫14(30.0%)、分散蓝1(73.0%)、直接蓝6(58.2%)、直接蓝15(87.5%)、酸性红26(81.0%)、直接红28(78.8%)、溶剂黄1(97.6%)、酸性紫49(99.8%)、直接棕95(80.5%)、分散橙11(96.7%)、直接黑38(52.0%)、碱性紫3(84.9%)、分散黄56(88.4%)、分散黄3(98.7%)、酸性红114(92.3%)、溶剂黄3(98.5%)、溶剂黄2(98.7%)、碱性蓝26(96.5%)、分散黄23(97.8%)、分散橙149(95.8%)、分散黄7(94.3%)、分散红151(98.4%)均购于德国Dr.Ehrenstorfer公司。
实际样品:239个牙膏样品和88个漱口水样品均由南京海关工业产品检测中心提供。
1.2 标准溶液的配制
标准储备溶液:根据各标准品纯度换算,分别准确称取一定量的23种致癌染料标准品,分别用乙醇溶解并定容至10 mL,配制成质量浓度均为1 000 mg/L的单一标准储备液,于4 ℃冷藏保存。
标准工作溶液:分别移取一定体积上述标准储备溶液,配制成混合标准溶液(分散蓝1、直接蓝6、直接蓝15、直接棕95、分散橙11、直接黑38和分散黄56的质量浓度为200 mg/L;碱性红9、酸性红26、直接红28和酸性红114的质量浓度为100 mg/L;碱性紫14、溶剂黄1、分散黄3、溶剂黄2、碱性蓝26、分散橙149、分散黄7和分散红151的质量浓度为50 mg/L;酸性紫49、碱性紫3、溶剂黄3和分散黄23的质量浓度为20 mg/L ),根据需要用乙醇稀释配制成系列标准工作溶液。
1.3 样品前处理
1.3.1 漱口水称取1.0 g(精确至0.01 g)样品,置于10 mL容量瓶中,用乙醇溶解并定容至刻度,混匀,取部分样品待测。
1.3.2 牙膏称取1.0 g(精确至0.01 g)样品,置于50 mL离心管中,加入适量水和石英砂,涡旋混合均匀,置于真空干燥器中75 ℃烘干后,加入10 mL乙醇,50 ℃超声30 min,以8 000 r/min离心3 min后,取部分上清液待测。
1.4 色谱条件
色谱柱:ZORBAX Eclipse XDB-Phenyl(150 mm×4.6 mm×5 μm);流动相:A为2.5 mmol/L磷酸二氢四丁基铵水溶液(pH 7.5),B为甲醇-乙腈(体积比50∶50)。梯度洗脱程序:0.0~15.0 min,15%~40%B;15.0~17.0 min,40%B;17.0~32.0 min,40%~85%B。流速:1.5 mL/min;进样体积:10 μL;柱温:40 ℃;检测波长:400、500、600 nm。
2 结果与讨论
2.1 混合标准溶液分组
配制染料混合标准溶液时,应考虑色谱分离、阴离子和阳离子化合物不能混合以及标准品互含等情况,需将混合标准溶液进行分组配制[21]。本实验配制了23种致癌染料的混合标准溶液(质量浓度见“1.2”),于配制当天与各物质相同浓度单标溶液的色谱行为进行比较,之后将混合标准溶液与单标溶液于4 ℃下避光保存,分别于第7、14、21、28、35 d对上述标准溶液进行测定。结果表明,混合标准溶液中23种致癌染料的保留时间与单标一致,各组分的峰面积基本相当,不同储存时间下混合标准溶液中23种致癌染料的峰面积与单标溶液峰面积的比值为92.5%~102%,因此推测所配制的混合标准溶液可在4 ℃下避光保存35 d。
2.2 色谱柱的选择
色谱柱是色谱分离体系的心脏,因此对色谱柱进行了选择。由于反相色谱柱柱效高,分离能力强,保留机理清楚,在液相色谱分离模式中应用最为广泛,本实验比较了2种常用反相色谱柱:C18柱(150 mm×4.6 mm×5 μm)和苯基柱(150 mm×4.6 mm×5 μm)对23种致癌染料的分离效果。结果显示,大部分待测物在C18柱上能够得到有效分离,但酸性紫49和直接棕95的分离效果不佳,而23种待测物在苯基柱上均能达到有效分离。因此,本研究最终选择苯基柱为分析色谱柱。
2.3 流动相的优化
23种致癌染料中包含多种以阴离子形式存在的化合物,如以水作为水相流动相,由于大分子结构空间位阻,使得这些致癌染料在反相色谱柱上保留较差甚至无保留。阳离子对试剂可增加阴离子型化合物与固定相的结合力,因此实验比较了带不同大小基团的阳离子对试剂(乙酸铵、乙酸二正己基铵、四丁基氟化铵和磷酸二氢四丁基铵)对23种致癌染料保留的影响,水溶液浓度均为2.5 mmol/L。结果表明,随着离子对试剂所带基团的增大,阴离子型致癌染料的保留随之增强,其中乙酸铵的效果最小,磷酸二氢四丁基铵的效果最明显,且不影响其它类型致癌染料的分离。
此外,考察了水相流动相的pH值对23种致癌染料色谱分离的影响,结果显示,当pH值为7.0~8.0时,23种待测组分均有较高的响应和较好的色谱峰形,因此选择pH 7.5的水相流动相。实验考察了分别以甲醇和乙腈作为有机流动相时的分离效果,结果表明乙腈不能使23种致癌染料得到有效分离,而采用甲醇时23种致癌染料的分析时间较长,峰形较差。并且由于甲醇和乙腈对23种致癌染料的洗脱能力不同,导致一些染料的出峰顺序发生改变。进一步比较了不同比例的甲醇-乙腈混合溶液作为有机流动相对待测物分离效果的影响,发现甲醇-乙腈(体积比50∶50)的分离效果最佳,因此选择2.5 mmol/L磷酸二氢四丁基铵水溶液(pH 7.5)和甲醇-乙腈(体积比50∶50)为流动相。优化色谱条件下,23种致癌染料混合标准溶液的液相色谱图见图1,可在32 min内完成色谱分离。
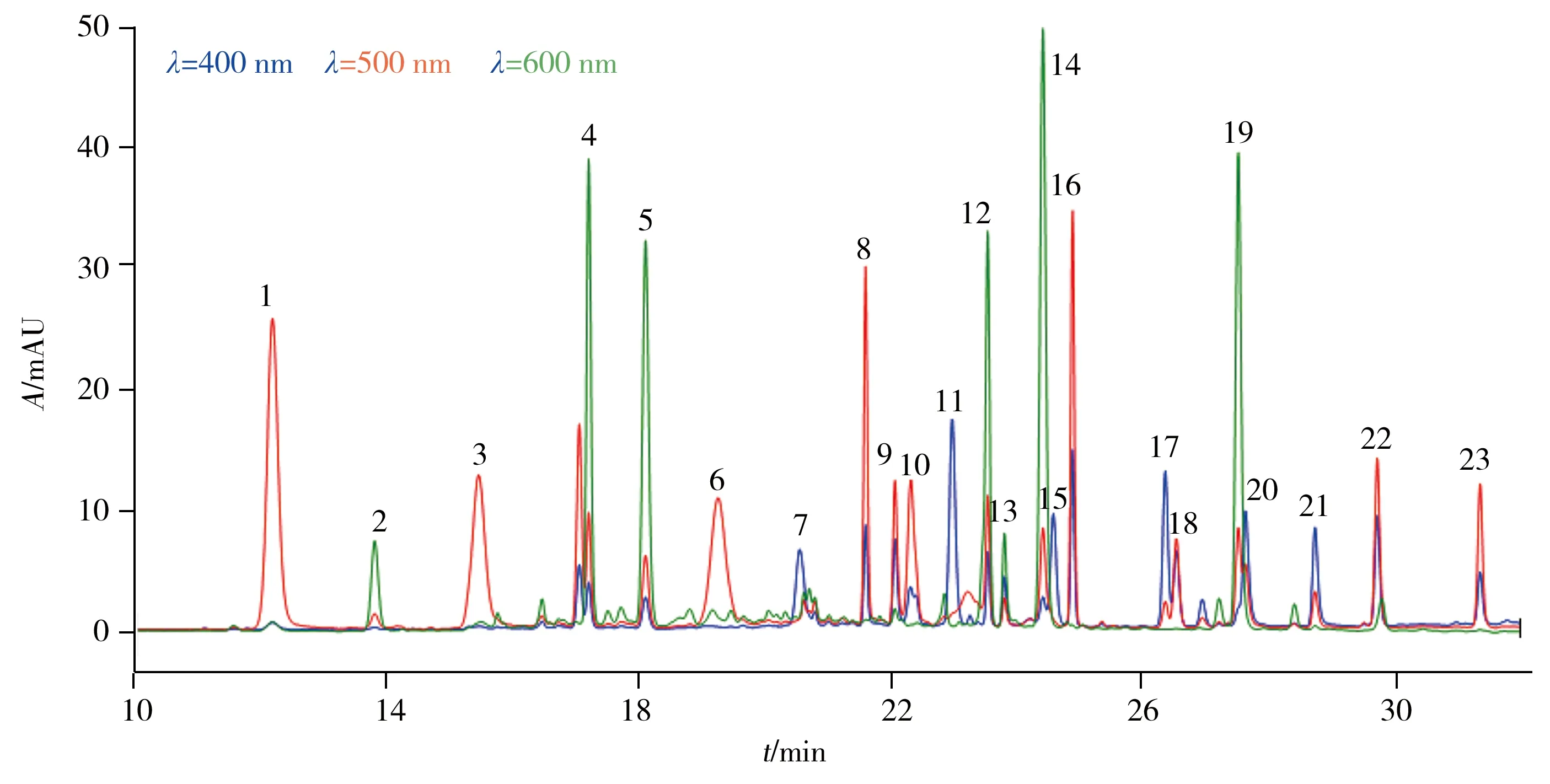
图1 23种致癌染料混合标准溶液的液相色谱图
2.4 紫外检测波长的选择
采用三维(3D)检测模式,分别对23种致癌染料的单标溶液在190~800 nm范围内进行紫外扫描,得到23种致癌染料的最大吸收波长。结合检测灵敏度及工作效率,本实验采用400 nm、500 nm和600 nm对23种致癌染料进行扫描测定。
2.5 前处理条件的优化
2.5.1 烘干条件的优化牙膏中含有羟甲基纤维素、羟乙基纤维素、鹿角果胶或黄原胶等增稠剂,这些增稠剂易溶于水,不溶于纯有机溶剂。牙膏中还含大量水分,如直接用有机试剂提取,部分增稠剂会溶于有机试剂与水的混合液中,导致进样溶液严重堵塞色谱柱,因此需对样品进行烘干处理。样品烘干前加入适量石英砂混匀,可增加样品与空气的接触面,缩短烘干时间,也可防止样品烘干后结块影响提取。样品水分的常用烘干温度为75 ℃和105 ℃,经试验可知,采用普通烘箱105 ℃烘干时,直接蓝15等不稳定染料会分解;当烘干温度为75 ℃,烘干时间较长,影响工作效率。真空干燥器可在真空状态下降低水的沸点,适用于热敏性物质的干燥,因此,本实验采用真空干燥器75 ℃烘干牙膏样品。
2.5.2 超声条件的优化23种致癌染料的极性分布较宽,其中直接黑38、直接蓝6、直接蓝15、直接棕95、直接红28等离子型致癌染料不溶于多数有机溶剂,但在乙醇中有较好的溶解性,因此选择乙醇作为提取溶剂。本实验采用超声萃取对牙膏样品进行提取,升高超声温度、延长超声时间和提高超声频率均能促进化合物的溶出和溶解,但温度过高会使不稳定化合物分解,超声时间过长会影响工作效率,超声频率过高会导致化合物结构发生变化。分别考察了在不同的超声温度(30、40、50、60 ℃)、超声时间(10、20、30、40 min)和超声频率(84、168、252、336、420 W)下,乙醇对同一自制阳性牙膏样品(待测组分的添加量均为100 mg/kg)的提取情况,结果见表1。由表1可知,当超声温度为50 ℃、超声时间为30 min和超声频率为252 W时,23种致癌染料的提取总量达最大值。因此,牙膏样品采用超声温度50 ℃、超声时间30 min和超声频率252 W为前处理条件。
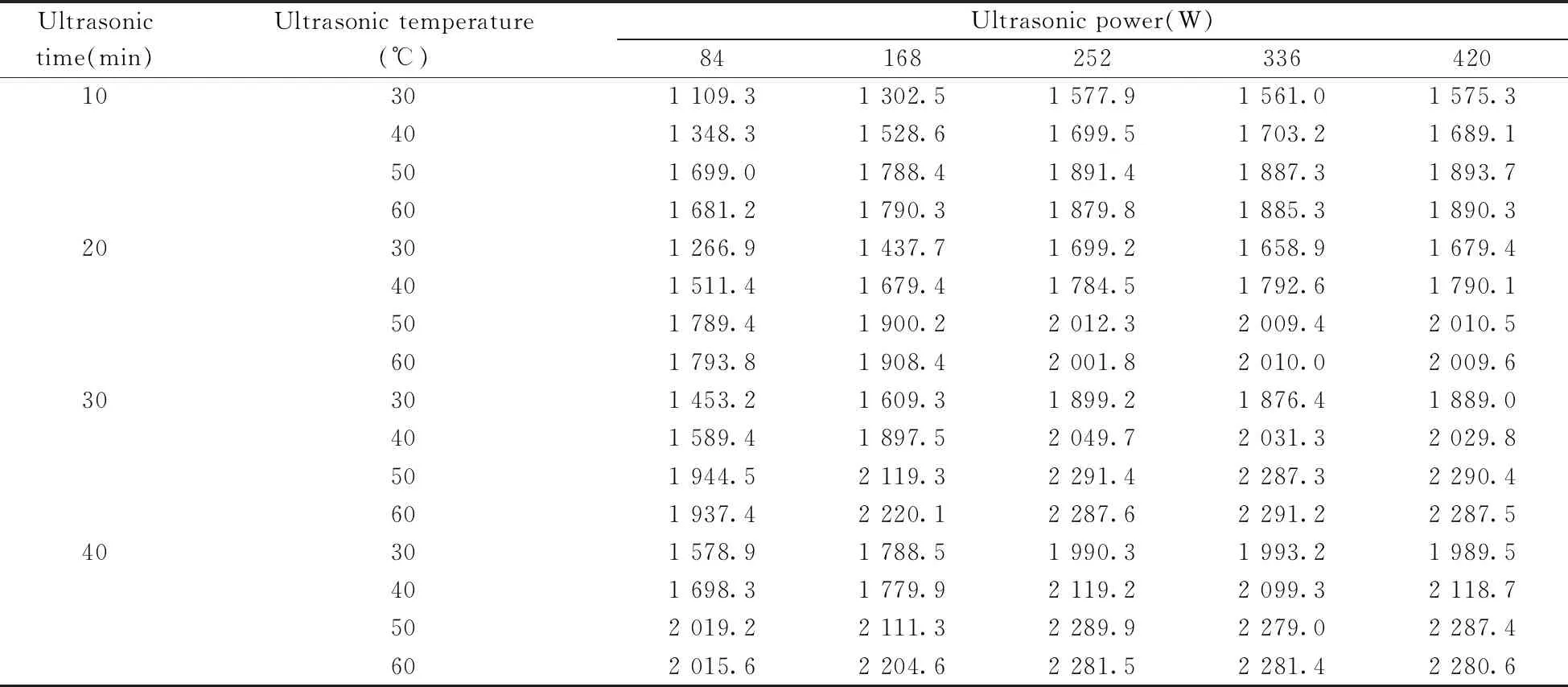
表1 不同超声温度、超声时间和超声频率下23种致癌染料的提取总量
2.6 方法学评价
2.6.1 线性范围、检出限与定量下限将“1.2”的混合标准溶液用乙醇逐级稀释,采用本方法进行测定,以各组分的峰面积(y)对其质量浓度(x,mg·L-1)进行线性回归,得到线性回归方程。结果表明,23种致癌染料在对应的质量浓度范围内线性关系良好,相关系数(r2)均不小于0.999 1。在阴性样品中添加不同浓度的待测组分,根据3倍和10倍信噪比分别计算检出限(LOD,S/N=3)和定量下限(LOQ,S/N=10),得到23种致癌染料的LOD为0.06~0.66 mg/L,LOQ为0.18~1.98 mg/L(见表2),能够满足目前的检测要求。

表2 23种致癌染料的保留时间、检测波长、线性关系、检出限与定量下限
2.6.2 回收率及相对标准偏差以阴性牙膏和漱口水样品为基质,分别进行3个不同加标水平的回收实验,每个加标水平作6次平行,按照“1.3”进行样品前处理,在“1.4”色谱条件下测定,计算得到23种致癌染料的平均回收率和相对标准偏差(RSD)。由表3可知,23种致癌染料在牙膏和漱口水中的平均回收率分别为91.2%~104%和92.0%~103%,RSD分别为2.5%~5.8%和2.3%~5.5%,表明该方法准确度好、精密度高,适用于牙膏和漱口水中致癌染料的测定。
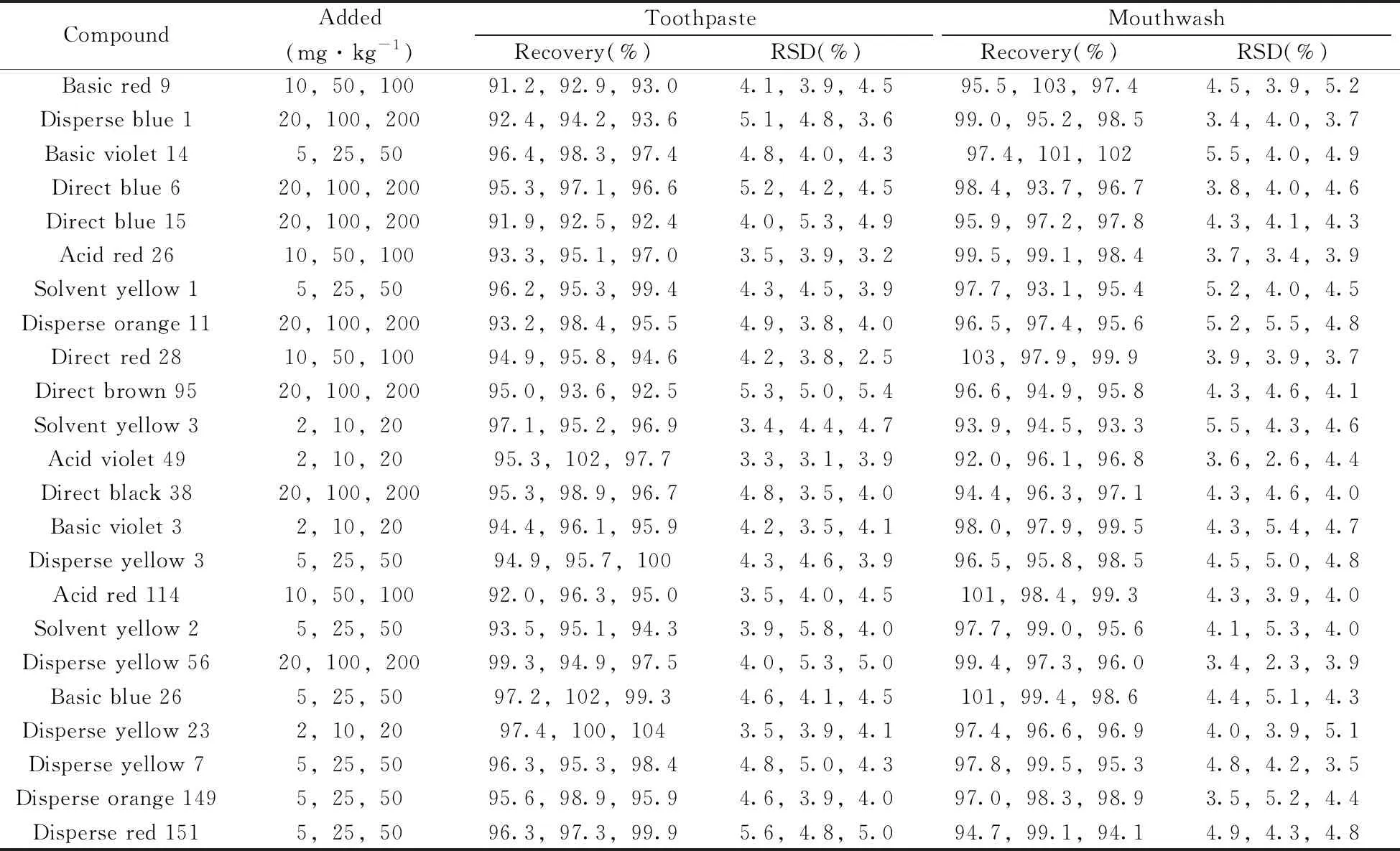
表3 阴性样品中23种致癌染料的回收率及相对标准偏差(n=6)
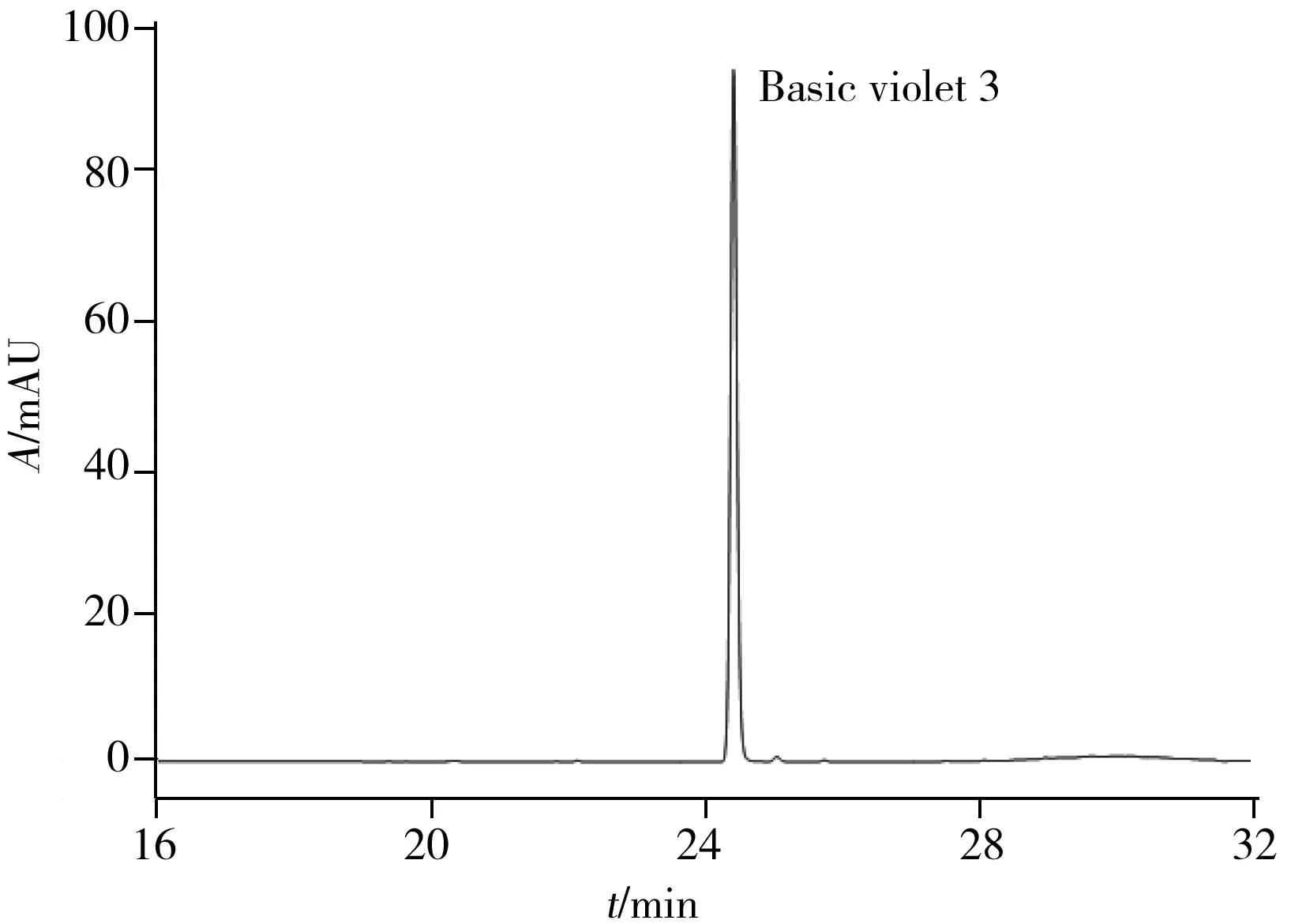
图2 阳性样品的色谱图
2.7 实际样品检测
应用本方法分别对239个牙膏和88个漱口水样品进行测定,其中1个牙膏样品检出碱性紫3,含量为53.4 mg/kg,色谱图见图2。
3 结 论
本文建立了高效液相色谱测定牙膏和漱口水中23种致癌染料的方法,并对色谱检测条件和牙膏样品前处理方法进行了优化。结果表明,该方法操作简单,线性关系良好,重复性好,具有较高的准确度和灵敏度,适用于各种牙膏和漱口水中致癌染料的检测,能够满足国内外对致癌染料残留限量的要求。
猜你喜欢
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
农家参谋(2021年10期)2021-12-05
中老年保健(2021年12期)2021-11-30
科技创新导报(2020年5期)2020-06-11
恋爱婚姻家庭·养生版(2019年2期)2019-02-26
祝您健康(2018年12期)2018-11-27
科教导刊(2017年26期)2017-11-07
科技与创新(2015年17期)2015-09-11
科技与创新(2014年12期)2014-08-28
