
Dysferlinopathy早期误诊为多发性肌炎一例
2021-02-25 05:23崔文帅任洁
临床内科杂志 2021年1期
崔文帅 任洁
患者,女,27岁,因“双下肢无力2年,加重1年”于2019年3月1日入院。入院前2年患者无明显诱因出现双下肢无力,近端较重,蹲下起身困难,未予重视。1年前上述症状加重,步行1 km自觉乏力明显,并逐渐出现双上肢抬举困难。病程中患者无发热、胸闷、关节肿痛等不适。为求进一步治疗来我院。患者既往身体健康,否认家族中类似病史及风湿性疾病病史。入院体格检查:T 36.7℃,P 75次/分,R 16次/分,Bp 125/75 mmHg,步态不稳,全身无皮疹,心肺腹检查无明显异常;四肢关节无肿胀压痛,活动正常,无肌痛及肌压痛,双上肢、双下肢肌力Ⅲ级,肌张力正常;生理反射正常存在,病理反射未引出。入院后完善相关检查:ALT 148.0 U/L(7.0~40.0 U/L,括号内为正常参考值范围,以下相同),AST 198.0 U/L(13.0~35.0 U/L),血清肌酸激酶(CK)6 640.0 U/L(26.0~174.0 U/L),血清肌酸激酶同工酶(CK-MB)186.0 U/L(3.0~25.0 U/L),乳酸脱氢酶(LDH)650.0 U/L(109.0~245.0 U/L),超敏C反应蛋白(hs-CRP)1.46 mg/L(0~8.00 mg/L),红细胞沉降率(ESR)53 mm/h(0~20 mm/h),抗核抗体(ANA)核颗粒型1∶100(+)。免疫球蛋白IgA、IgG、IgM、补体C3、C4、甲状腺功能、肿瘤标志物、性激素六项、类风湿因子、抗环瓜氨酸肽(CCP)抗体、血管炎五项、抗心磷脂抗体检查结果均为阴性。肌电图检查结果示肌源性损害。肌肉组织病理活检结果示(右小腿肌肉)镜下见肌纤维变性,间质见淋巴细胞等炎细胞浸润,小血管增生,符合肌炎特征。诊断考虑多发性肌炎(PM),予患者甲强龙80 mg静脉滴注3天、环磷酰胺(CTX)0.6 g静脉滴注2天抑制免疫及护胃、保肝、改善微循环、营养神经等综合治疗后,患者诉肌无力好转,出院前查CK 2 052 U/L,CK-MB 53 U/L。出院后患者服用美卓乐40 mg每日1次并逐渐减量,且在当地医院应用环磷酰胺每月1.2 g治疗。2019年8月患者旅游后出现双下肢肌无力加重,于当地医院复查CK 3 425 U/L、CK-MB 235 U/L。予甲强龙500 mg冲击治疗连续3天,后予美卓乐40 mg口服,患者肌无力无明显好转,复查CK 6 815 U/L。为进一步治疗患者再次就诊于我院,考虑其对激素及免疫抑制剂治疗效果欠佳,再次行肌肉组织病理活检结果显示:1.常规及酶组织化学染色:肌纤维大小不均,可见散在、小簇分布的小角状、小圆形萎缩肌纤维;并可见个别坏死、吞噬肌纤维(图1D);2.免疫组织化学(IHC)染色:Dysferlin肌膜肌纤维着色欠佳,部分缺失(图1A、1B、1C),主要组织相容性复合体(MHC)-1肌膜弱(+);膜攻击复合体(MAC)肌膜弱(+);CD68肌纤维间隙散在(+);CD79a(-);CD8、CD4个别(+);CD34毛细血管密度未见下降;I型干扰素诱导蛋白(MxA)(-);α-sarco、β-sarco、γ-sarco、δ-sarco、Dys-N、Dys-C、Dys-R肌纤维膜均匀表达;caveolin-3肌纤维均匀表达;3.电镜超微病理:肌细胞肌原纤维排列基本规则,部分肌原纤维Z线灶性紊乱,局部见肌原纤维撕裂、多灶状溶解,肌膜皱缩;肌细胞轻度大小不均、间隙无明显增宽,局部胶原纤维轻度增生,未见明显炎细胞浸润,毛细血管基膜未见明显增厚。镜下送检组织病理改变考虑dysferlinopathy,建议患者行基因检查明确诊断。患者及其父母完善基因检测,结果显示其存在DYSF基因突变,确诊为dysferlinopathy。
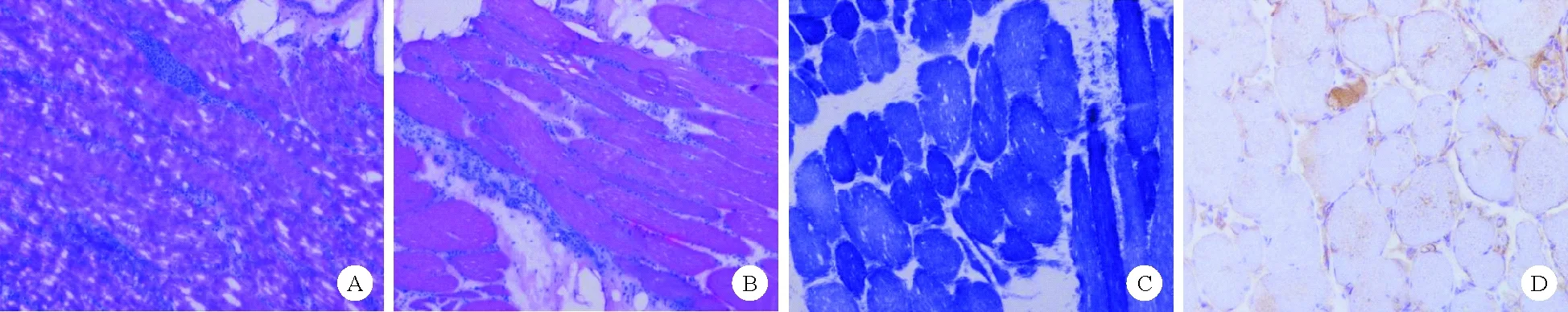
图1 患者肌肉组织病理活检结果[A、B:肌间纤维结缔组织未见明显增生,少许脂肪细胞浸润;肌间毛细血管扩张,未见明显淋巴细胞浸润;肌纤维大小不均,直径10~100 μm,可见散在、小簇分布的小角状、小圆形萎缩肌纤维,未见束周肌萎缩;可见核聚集;可见内核肌纤维,比例3%~10%;可见个别坏死、吞噬肌纤维,未见再生肌纤维;苏木素-伊红(HE)染色,×40;C:两型肌纤维分型欠佳;还原性辅酶(NADH)染色,×40;D:Dysferlin肌膜肌纤维着色欠佳,部分缺失;苏木素染色,×40]
讨 论
Dysferlinopathy是一组由DYSF基因突变引起的高度异质性的常染色体隐性遗传性肌肉变性疾病。根据首先受累肌群的不同主要分为以下肢后部近端肌及骨盆肌无力为主的肢带型肌营养不良2B型(LGMD2B)和以小腿后群肌肉无力为主的Miyoshi远端型肌营养不良(MM)两种类型。各类型的共同特点为患者体内均存在DYSF基因突变,导致dysferlin蛋白完全或部分缺失[1]。Dysferlin蛋白为ferlin家族成员之一,在参与肌细胞膜损伤的修复及内皮细胞粘附和血管生成的过程中发挥着至关重要的作用[2]。正常情况下,当骨骼肌质膜损伤后,dysferlin蛋白作为一种可感受Ca2+的关键感受器,和其他修复蛋白(caveolin和calpain等)通过以Ca2+依赖的方式与磷脂结合,使大量的Ca2+进入骨骼肌细胞质,引起损伤部位的囊泡积聚和肌膜融合,从而修复受损伤的肌膜。Dysferlin蛋白可在多种组织中表达,但主要存在于骨骼肌和心肌细胞膜内面或胞质囊泡内,也可表达于单核细胞[3]。DYSF基因突变可导致不同程度的dysferlin蛋白表达减低、缺失或功能障碍,引起膜修复过程受损;另一方面,肌膜内损伤相关分子模型(DAMPs)如热休克蛋白(HSP)、尿酸分子、高迁移率族蛋白、ATP和自由基等[4]与Toll样受体结合,激活核因子(NF)-κB、补体、单核细胞[5]、巨噬细胞及炎症因子[6],进一步加剧肌膜的损伤,肌膜失去自我修复能力,引起肌纤维坏死和炎性反应,肌细胞逐渐被脂肪和纤维组织替代[7]或在早期出现骨骼肌和脂肪组织脂质代谢的重新编辑[8-9],从而出现渐进性的肌无力、肌萎缩等症状。由于dysferlinopathy和PM在临床特征和组织学上都存在较高的相似性,因此在实际临床工作中,两者之间极易被误诊。
本例患者为青年女性,起病隐匿,主要症状为双下肢近端肌无力进行性加重,CK水平明显升高,肌电图检查结果示肌源性损害,初次肌肉组织病理活检结果提示肌炎性改变,诊断为PM。予激素联合CTX治疗后患者肌无力症状短时缓解,CK水平下降,但劳累后肌无力症状再发加重,予激素冲击治疗效果欠佳,后再次行肌肉组织病理活检并于外院完善基因检测,确诊为dysferlinopathy。由于患者不愿透漏个人及双亲基因检测突变位点结果,暂无法明确dysferlinopathy属于何种亚类,但根据其病史、发病特点及免疫组化、组织病理活检等检查结果综合考虑LGMD2B可能性较大。
LGMD2B多见于青中年人,以女性多见。主要以近端骨盆肌肉和大腿后肌肉无力为首发症状,受累肌肉主要为四肢近端,下肢为臀肌、阔筋膜张肌、大收肌、腘绳肌。随着疾病的逐渐进展,可累及肩胛带肌和上肢肌,但较少累及面肌、颈肌、手部肌肉等部位。早期也可表现为无症状高肌酸激酶血症和转氨酶升高,CK水平高达20 000 U/L。肌肉病理改变多样,其中最明显的特征为受累肌出现明显的坏死。有研究报道约50%患者的肌内膜和血管周围有CD4+T细胞和巨噬细胞浸润,容易被误诊为PM。肌电图检查结果可显示肌源性损伤,而组织病理学检查结果主要为dysferlin蛋白的缺失。
临床上dysferlinopathy早期误诊为PM较为常见,需要通过IHC和(或)蛋白质免疫印迹法(Western blot)分析鉴别两者。IHC或Western blot分析结果表明,dysferlinopathy患者MHC-Ⅰ呈阴性或弱阳性表达,肌纤维膜Dysferlin蛋白表达降低或缺失,通常认为Dysferlin蛋白下降到正常值20%以下时可诊断原发性dysferlinopathy。而PM患者结果却相反[10]。因Dysferlin蛋白在多种组织中均可表达,因此Western blot较IHC敏感。在疾病早期,联合两种检查可提高对该病诊断的敏感性,而基因筛查则是确诊该病必不可少的手段。
临床上无论是首先怀疑PM或dysferlinopathy(LGMD2B和MM),早期的正确诊断直接决定着患者预后。大多数PM患者对激素或免疫抑制剂较为敏感,早期使用激素或联合免疫抑制剂治疗可阻止疾病进展,恢复肌力。而dysferlinopathy患者却恰恰相反。因此,对于怀疑早期肌无力或高CK血症的患者,排除功能性因素外,当使用激素或免疫抑制剂治疗后症状无明显改善时,及时选择IHC或Western blot联合二代基因检测很有必要。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
当代医药论丛(2021年24期)2022-01-20
中国畜牧杂志(2021年10期)2021-12-03
临床误诊误治(2021年3期)2021-03-30
国际放射医学核医学杂志(2021年10期)2021-02-28
中华养生保健(2021年18期)2021-02-13
灌篮(2020年36期)2020-05-16
中国医药科学(2017年9期)2017-08-04
蚌埠医学院学报(2016年5期)2016-12-16
放射学实践(2016年6期)2016-12-15
