
8个番石榴品种的转录组测序
2021-02-12 02:51张丽梅陈钟佃张朝坤魏秀清许家辉
福建农业学报 2021年11期
张丽梅,陈钟佃,张朝坤,魏秀清,许家辉
(1. 福建省农业科学院果树研究所, 福建 福州 350013;2. 福建省农业科学院农业生态研究所, 福建 福州 350013;3. 福建省漳州市农业科学研究所, 福建 漳州 363000)
0 引言
【研究意义】番石榴(Psidium guajavaLinn.)原产于南美洲大陆,广泛栽植于热带亚热带地区,在我国华南各地也均有栽培,是重要的药食同源植物。番石榴营养丰富,风味独特, 在水果市场特别是饮料市场上占有重要的地位,近年来市场对番石榴果汁的需求量也在快速增长。同时,番石榴作为传统药用植物在美洲、非洲和亚洲等地被用于治疗腹泻、痢疾和皮炎等疾病[1]。番石榴中大量的酚类、类胡萝卜素、萜类和三萜类等已经被证实具有抗氧化[2]、保肝[3,4]、抗过敏[5]、抗菌[6,7]、抗细胞毒性[8,9]、抗龋齿[10,11]、抗糖尿病[2,12]、抗炎症[13]等用途,并且已经广泛应用到临床。【前人研究进展】目前对于番石榴的研究多集中在采后生理[14-17]、内含物萃取[18-20]、药用价值[21]以及果汁产品的开发[22,23]等方面。【本研究切入点】关于番石榴植株本身营养成分富集和有效药用成分积累的机理和相关功能基因的研究还鲜见报道。在美国国立生物技术信息中心(National Center for Biotechnology Information)数据库中番石榴仅有132个基因和84个蛋白质信息,这极大地阻碍了番石榴生物技术相关研究的开展。因此,通过高通量测序平台获得番石榴转录组信息,可以为番石榴后续功能基因的鉴定提供参考。【拟解决的关键问题】本试验通过Illumina HiSeq 2500高通量测序平台对8个番石榴品种进行转录组测序,利用公共数据库对测序结果进行生物信息学分析,旨在为番石榴酚类、萜类、多糖类等生物合成途径功能基因的开发奠定基础。利用高通量测序分析8个番石榴种质资源之间的基因表达情况,通过生物信息学分析找寻其内在联系,同时为番石榴分子机制相关研究提供丰富的数据资源。
1 材料与方法
1.1 试验材料
本试验8个番石榴(Psidium guajavaLinn.)品种果实来源于福建省农业科学院果树研究所,包括红心(HX)、彩虹(CH)、紫红(ZH)、水晶(SJ)、秀珍(XZ)、彩叶变(CYB)、彩叶(CY)和紫星(ZX)等。8~9分成熟果实采摘,洗净晾干切块装入冻存管,投入液氮保存备用。液氮保存的样品交由杭州祥音生物医药科技有限公司进行文库构建及测序。
1.2 番石榴文库构建及转录组测序
采用Trizol 法分别提取总RNA,分别采用Nanodrop、Qubit 2.0、Aglient 2100方法检测RNA样品的纯度、浓度和完整性等。用带有Oligo(dT)的磁珠富集真核生物mRNA,加入Fragmentation Buffer将mRNA进行随机打断,以mRNA为模板,用六碱基随机引物(Random hexamers)合成第一条cDNA链,加入缓冲液、dNTPs、RNase H和DNA polymerase I合成第二条cDNA链,利用AMPure XP beads纯化cDNA。纯化的双链cDNA再进行末端修复、加A尾并连接测序接头,AMPure XP beads进行片段大小选择,最后通过PCR富集得到cDNA文库。
通过Illumina HiSeq 2500测序平台和PE125测序方法对番石榴构建转录组文库后进行测序,测序得到的原始图像数据经base calling 转化为原始数据(Raw reads),使用FastQC软件对原始数据进行质量评估。Trimmomatic软件[24]质量剪切获得Clean数据。最后利用Trinity软件[25]de novo组装成转录本,统计所得转录本的各项信息。
1.3 番石榴基因功能注释
使用NCBI Blast+软件[26]将转录本与保守结构域数据库CDD (Conserved domain database)、真核生物蛋白质同源数据库 KOG(Karyotic ortholog groups)、蛋白质直系同源数据库COG(Cluster of orthologous groups)、非冗余蛋白数据库NR(Non-redundant protein database)、核酸序列数据库NT (NCBI nucleotide sequences)、蛋白质结构域数据库 PFAM(Protein family database)、蛋白质序列数据库Swiss-Prot (A manually annotated and reviewed protein sequence database)、Swiss-Prot增补本TrEMBL数据库等多个数据库进行比对,得到转录本的注释信息。根据转录本与Swissprot、TrEMBL的注释结果得到基因本体论数据库GO (Gene ontology) 功能注释信息。利用KAAS软件得到转录本在东京基因与基金组百科全书数据库KEGG (Kyoto encyclopedia of genes and genomes)注释信息。根据转录本与数据库Blast比对结果和TransDecoder软件进行CDS预测。
1.4 番石榴RNA-seq测序质量评估
使用Bowtie 2软件[27]将样本有效数据比对到拼接所得的转录本上,统计Mapping信息。利用RSeQC软件[28]根据比对结果进行冗余序列分析、插入片段分布等分析。BEDTools[29]进行均一性分布检查和基因覆盖率统计分析。
1.5 基因表达及基因富集分析
使用Salmon[30]和WGCNA[31]分别进行基因的表达量计算和基因共表达分析。基于样本的表达量矩阵进行样本比较分析等多方向的统计分析和探索。使用DESeq2[32]进行基因表达差异分析,对表达差异分析结果进行可视化。基于差异分析结果,绘制基因功能表达韦恩图、样本间距离热图,并进行主成分分析(Principle component analysis,PCA)。使用topGO进行GO富集分析。使用clusterProfiler[33]进行KEGG通路和KOG分类富集分析。
2 结果与分析
2.1 番石榴RNA-seq测序结果与数据组装
2.1.1 RNA-seq测序质量评估 8个品种的总碱基数为5184950713~8029343436 bp,平均7014638271 bp。Read均长为144~146 bp,平均145 bp;GC含量为49.98%~51.23%,平均50.48%;Q30为95.31%~95.96%,平均50.48%(表1)。可见,所获得的转录组质量较高。
2.1.2 RNA-seq测序数据组装 组装共获得283853条 Transcript。总序列信息达388691331 bp(0.39 Gb),平均长度1369 bp,Transcript的N50为2517 bp,N90为579 bp。其 中 长 度 为200~300 、300~500、500~1000、1000~2000 、≥2000 bp分别占23.31%、15.92%、16.20%、19.73%、24.83%。所得Transcript序列再次组装后得到126979 条 Unigene,Unigene总序列信息达97373299 bp(97.37 Mb),平均长度766 bp,各长度分布图如图1所示长度为200~300、300~500、500~1000 、1000~2000、≥2000 bp分别占42.81%、23.23%、14.56%、9.40%、9.99%。Unigene的N50为1654 bp,N90为267 bp,组装完整性较高。番石榴转录组测序结果的isoform分析表明100373个Unigene含有1个isoform,占比79.05%;含有2个isoforms的数量次之,共8109个Unigene,占比6.39%;含有3个及以上isoforms的Unigene共有18497 个,占比14.57%。
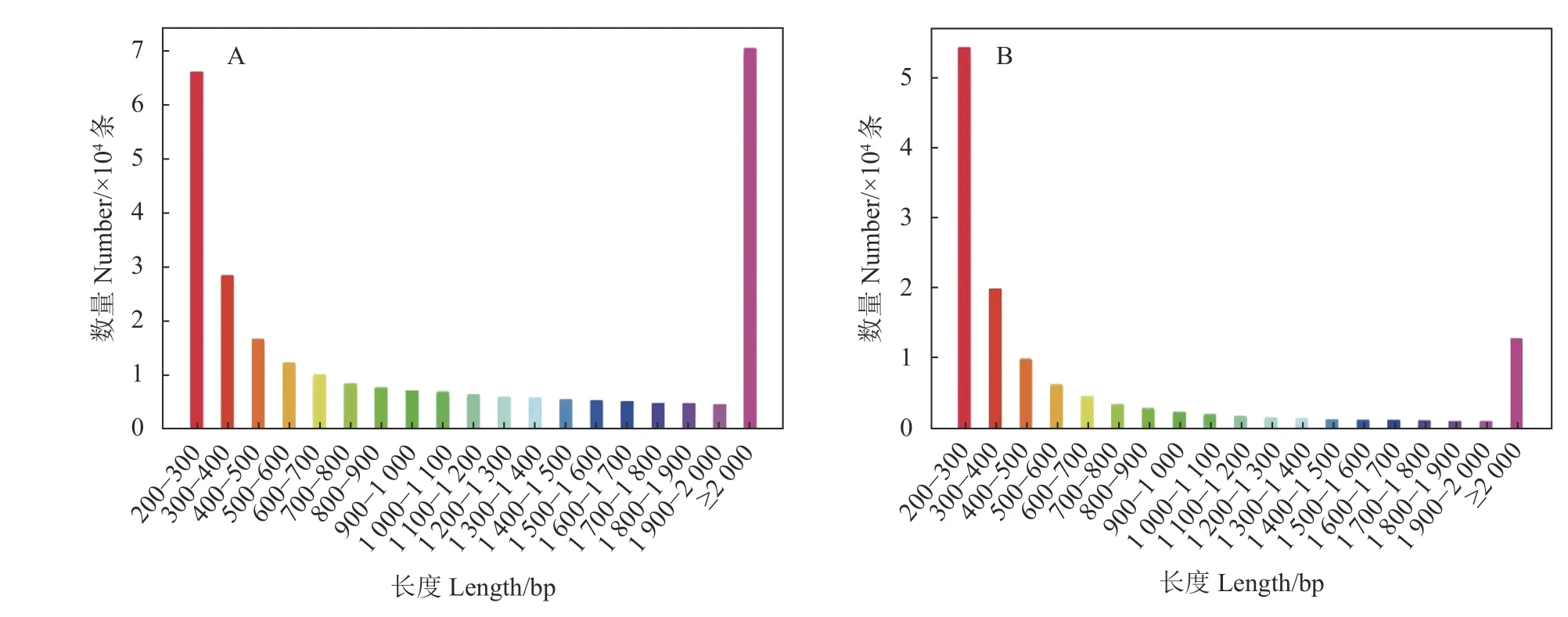
图1 Transcript(A)和Unigene(B)长度分布Fig. 1 Distributions of transcript length (A) and Unigene length (B)
2.1.3 SSR与SNP分析 8个番石榴品种含有SSR位点的Unigene共30265个,其中含有单碱基重复(p1)类型的Unigene最多,共15432个,占总数的50.99 %;双碱基重复(p2)类型的Unigene共7614个,三碱基重复(p3)类型的Unigene有3846个,四碱基重复(p4)类型的Unigene共314个,五碱基重复(p5)类型的Unigene共104个,六碱基重复(p6)类型的Unigene共68个。含有至少2个SSR位点的Unigene共2887个,含有至少2个位点且存在共用碱基的类型有172个。
8个番石榴品种发生单核苷酸变异的基因个数在42941~77098,发生片段插入或片段缺失的基因个数在13780~18435。其中HX发生变异数量最多,SJ发生变异数量最少。
2.2 番石榴Unigene功能注释
2.2.1 NR数据库比对注释 共有82283个Unigene被注释到NR数据库(图2),占所有基因数目的64.8%。NR功能注释匹配的物种中26007个Unigene(31.63%)与巨桉(Eucalyptus grandis)得到匹配,与番石榴同为桃金娘科(Myrtaceae Juss.)。其中E值小于1E-150 的Unigene有9066个(11.03%),E值介于1E-100 到1E-150 之间的Unigene有5516个(6.71%),E值介于1E-50 到1E-100 的Unigene有12782个(15.54%),E值介于1E-5到1E-50的Unigene有54905个(66.77%)。匹配序列相似度80%以上的Unigene有50798个(61.78%),相似度40%~80%的Unigene有30860个(37.53%),相似度低于40%的Unigene有625个(0.76%)。
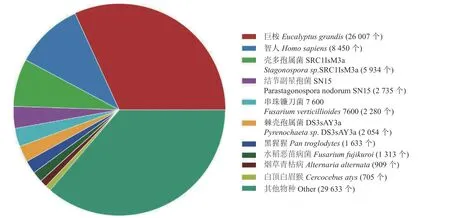
图2 番石榴Unigene NR数据库注释分析Fig. 2 Annotation of NR database on guava Unigenes
2.2.2 GO功能分类 GO功能分类注释结果(图3):21414个Unigene被分为3个本体71个功能组,共计787311个GO条目。其中222806个GO条目被分类到细胞组分的22个功能组中,细胞(55714个,18.62%)、细胞部分(55636个,18.59%)、细胞器(44023个,14.71%)和膜(31509个,10.53%)功能组中涉及的Unigene较多;185476个GO条目分类到22个分子功能组,其中结合活性(44261个,40.00%)和催化活性(34292个,30.99%)功能组中涉及的Unigene较多;374139个GO条目被分类到27个生物学过程功能组,细胞进程(51000个,13.51%)、单一生物进程(41616个,11.03%)以及代谢进程(40882个,10.83%)功能组中涉及的Unigene较多。
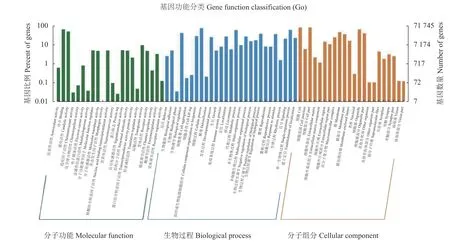
图3 番石榴Unigene Go分类Fig. 3 GO functional classification of guava Unigenes
2.2.3 KOG功能分类 将番石榴Unigene与KOG数据库进行比对(图4),并对其结果进行功能分类统计其中信号转导机制注释到最多,共计5577个,占比15.72%;其次是一般功能预测3726个,占比10.50%;以及翻译后修饰、蛋白折叠和分子伴侣3072个,占比8.66%。

图4 番石榴Unigene的KOG数据库分类Fig. 4 KOG classification of guava Unigenes
2.2.4 KEGG功能分类 番石榴7236个Unigene在KEGG数据库中的比对结果如图5所示。306个代谢途径中得到注释,其中核糖体途径(ko03010,Ribosome)被注释到500个Unigene,氨基酸生物合成途径(ko01230,Biosynthesis of amino acids)注释到264个Unigene,碳代谢途径(ko01200,Carbon metabolism)注释到260个Unigene。
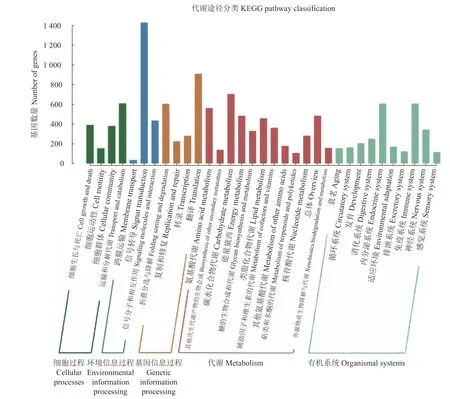
图5 番石榴Unigene的KEGG数据库分类Fig. 5 KEGG classification of guava Unigenes
2.3 番石榴PCA主成分与相关性及聚类分析
通过R包vegan构建番石榴8个品种间的PCA分析结果如图6所示。SJ和CYB在PCA 3D中直线距离均为最短,表明其主成分相似性最高;而品种CH和CY在两个3D图中距离均较远,表明其主成分之间相似性不高;在PCoA 3D中品种之间被分成4个区域,ZX、HX、XZ、和CH之间距离较近分布在一个区域,SJ和CYB分布在一侧,而品种CY和ZH距离前2个区域的品种距离较远,被划分到不同区域。
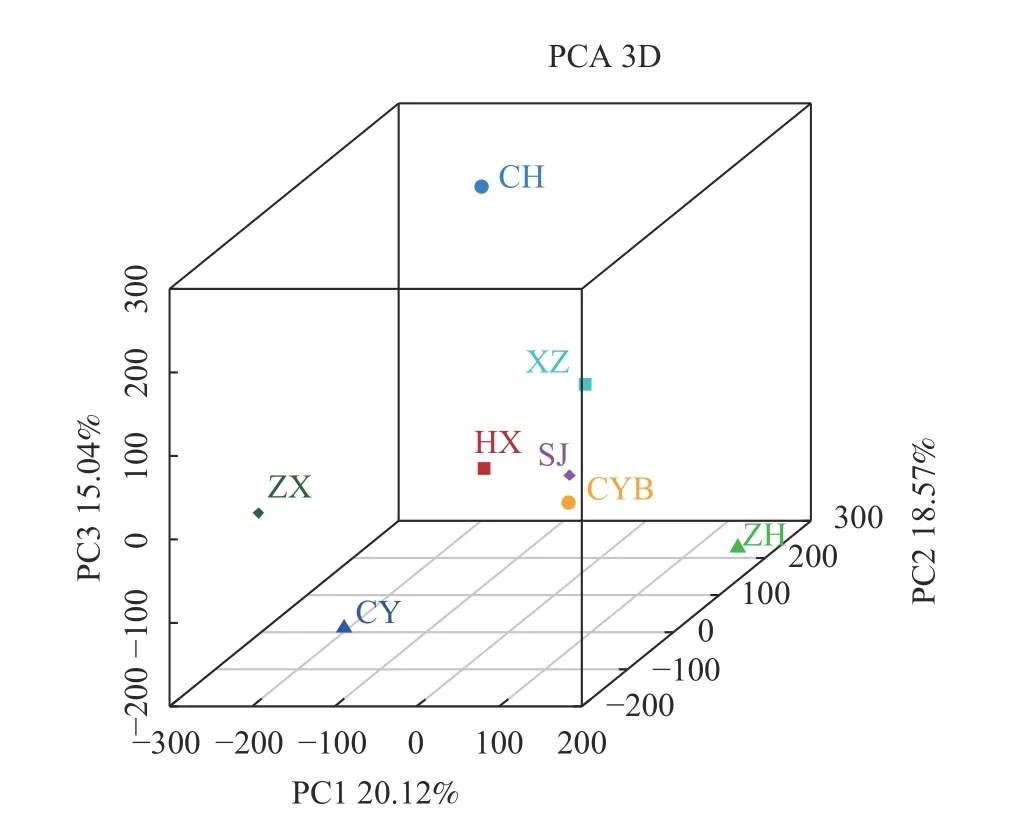
图6 番石榴PCA主成分分析Fig. 6 PCA on guava
使用统计算法bray curtis计算样本间距离,然后进行层次聚类(Hierarchical cluatering)分析,构建树状结构,得到树状关系形式用于可视化分析。通过R包gplots构建番石榴样本间距离热图(图7),8个样品间距离分布在0.21~0.52;其中SJ和CYB归为一类,两者间距离为0.3;其余6个品种归为一类,但品种ZH与所有品种的距离在0.37~0.49,距离较远;而HX、CH、XZ、ZX和CY等5个品种的距离在0.21~0.29,距离较近,相似度较高。
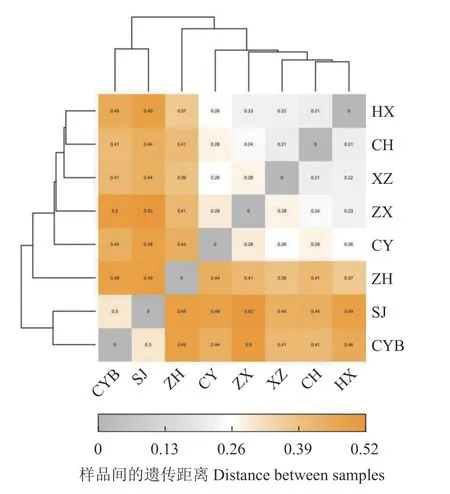
图7 样本遗传距离分析Fig. 7 Heat map of distance analysis on guava samples
2.4 番石榴品种间基因共表达分析
使用R包VennDiagram对8个番石榴品种共有的和独有的表达基因 (TPM > 0)的数目进行VENN图绘制(图8),直观的展现出样本中的表达基因数目组成相似性及重叠情况。8个番石榴品种含有27274个基因为共有的表达基因,而非重叠区表示不同样本之间特有的表达基因数。8个品种的特有表达基因数为1197(SJ)~5128(CH)。
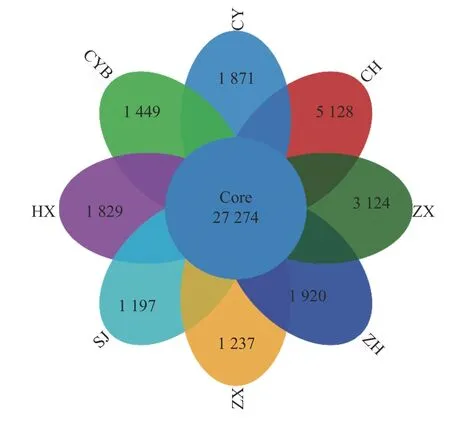
图8 番石榴基因共表达韦恩图Fig. 8 TPM Venn diagram of guava cultivars
2.5 番石榴8个品种间基因表达差异统计分析
8个番石榴品种进行基因差异表达分析结果(表2)表明:上调表达基因数为1994(CYB vs CY)~13355(CH vs HX),平均数为4451;下调表达基因数为1896(CH vs HX)~11471(SJ vs ZH)之间,平均数为6898;非差异基因数为109549(SJ vs ZH)~118012(CYB vs CY),平均数115629。KEGG富集途径分析表明:仅HX vs SJ未富集核糖体途径,其余组别均富集该途径;仅HX vs CH未富集碳代谢途径;仅ZH vs ZX及HX vs CH未富集氨基酸生物合成途径;仅HX vs SJ及ZH vs ZX富集淀粉和蔗糖代谢途径。
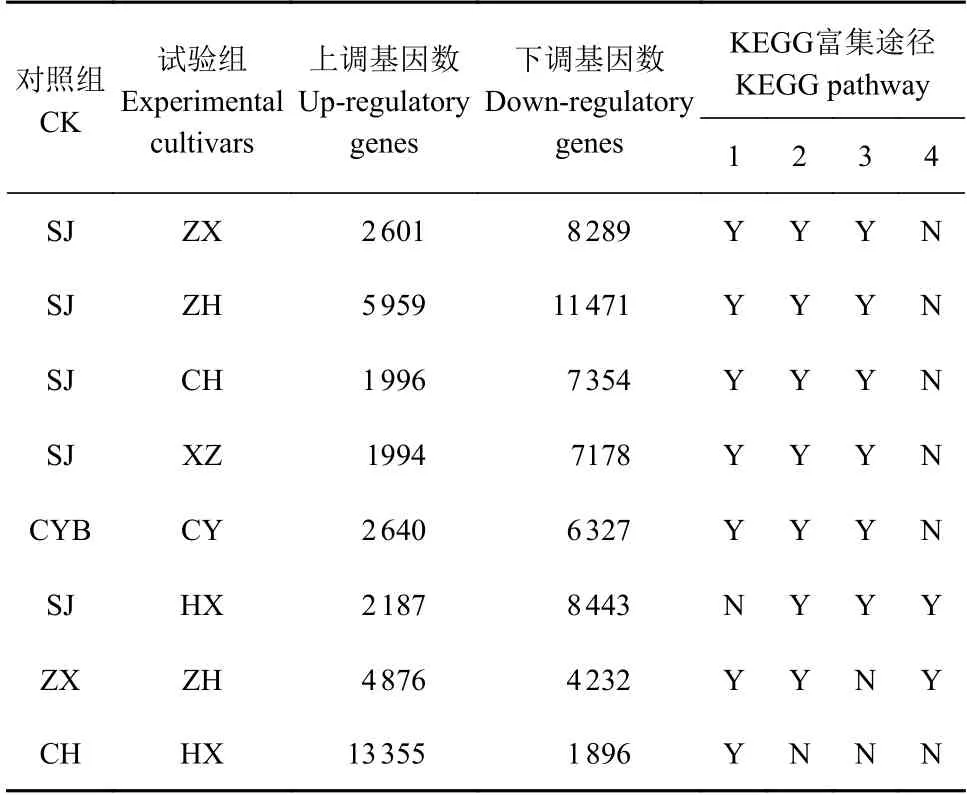
表2 番石榴8个品种间基因表达差异统计分析Table 2 Statistical analysis on gene expressions of 8 guava cultivars
3 讨论与结论
番石榴为桃金娘科常绿木本植物,具有较高的食用和药用价值。本研究较为系统地研究了番石榴的转录组,获得了大量的Clean data,其中Q30和GC含量分析结果均符合要求。此外,在Nr、KOG、GO、KEGG等数据库中,大量Unigene得到有效注释,组装的N50和N90参数分别为2517 bp和579 bp。此外,还获得了大量番石榴SSR和SNP标记。整体上,本研究所构建的转录组文库较为可靠和准确,具有较高的参考价值,可为后续研究提供科学参考。
转录组分析可为揭示植物品种间的特征差异形成机理提供重要的技术手段。目前,番石榴的转录组相关方面的研究较少[34]。本试验中,通过转录组分析发现水晶(SJ)和彩叶变(CYB)聚为一类,红心(HX)、彩虹(CH)、秀珍(XZ)、彩叶(CY)、紫星(ZX)归为一类,但品种紫红(ZH)与其他品种间距离较远。说明紫红品种的分子特征较为特殊,而水晶和彩叶变的亲缘关系较近,其余红心(HX)、彩虹(CH)、秀珍(XZ)、彩叶(CY)和紫星(ZX)的亲缘关系较近。聚类树状图与前期基于果实性状的研究结果较为一致[35],为番石榴种质资源分类提供了一定的参考。例如水晶(SJ)和彩叶变(CYB)的果皮颜色均为黄绿色;仅紫红(ZH)的果颜色为紫红色。在代谢途径差异方面,不同品种中富集的代谢途径并不完全一致,是导致品种果实间差异的重要原因,在后续分析中,仍需要进一步结合品种特性,关联转录组分析结果更加系统地揭示形成品种间性状差异的分子机理。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
中国果业信息(2021年12期)2021-12-02
科学导报(2021年29期)2021-06-03
科学之谜(2021年2期)2021-04-25
农村百事通(2021年3期)2021-04-16
科学导报(2020年54期)2020-09-09
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
科海故事博览·下旬刊(2019年6期)2019-04-16
饮食科学(2016年9期)2016-11-18
