
儿童线状IgA/IgG大疱性皮病一例
2021-02-06 12:06:52陈祚玺于长平施仲香周桂芝糜自豪张福仁
中国麻风皮肤病杂志 2021年2期
陈祚玺 于长平 王 娜 施仲香 周桂芝 糜自豪 张福仁
1山东大学齐鲁医学院,山东济南,250012;2山东第一医科大学附属皮肤病医院(山东省皮肤病医院,山东省皮肤病性病防治研究所),山东济南,250022
线状IgA大疱性皮病(linear IgA bullous dermatosis,LABD)是一种少见的以基底膜带上连续性线状IgA抗体沉积为特点的自身免疫性疱病[1],部分患者的基底膜带上同时有IgA抗体和IgG抗体沉积,此种则被称之为线状IgA/IgG大疱性皮病(linear IgA/IgG bullous dermatosis,LAGBD)[2],国内目前相关报道较少。我院诊治1例紫外线诱发难治性儿童LAGBD报道如下。
临床资料患儿,男,7岁。因躯干、四肢散发红斑、水疱伴瘙痒2个月来诊。2个月前患儿于泳池长时间“暴晒”后,当晚自觉暴露部位皮肤颜色稍红,偶有瘙痒,次日发现四肢散在红斑、水疱,以双侧手腕、小腿下1/3为著,伴瘙痒,后皮疹逐渐发展至躯干,同时上腭出现水疱,破溃后形成糜烂面。曾于外院诊断“线状IgA大疱病”,给予糖皮质激素(最高至泼尼松相当量75 mg/d,共28天)、人免疫球蛋白(5 g/d,共4天)、柳氮磺吡啶(最高至1 g/d,共15天)。原有皮疹部分消退,仍有新发水疱。既往患儿多次日晒后出现暴露部位变红,自行消退慢。否认近期感染史、服药史、肿瘤史等病史。
体格检查:系统检查未见异常。皮肤科检查(图1):皮疹散在分布于全身,口周、背部、四肢更多见,基本损害为正常皮肤或红斑基础上大小不一紧张性水疱、大疱,疱壁厚,疱液清亮或淡黄色,尼氏征阴性。部分水疱破溃结痂。头皮糜烂面可见渗液,皮疹占体表面积的10%;口腔软腭可见一处水疱及一处1 cm×2 cm糜烂面,周围有红晕;外阴及肛门处皮肤未见水疱和糜烂。
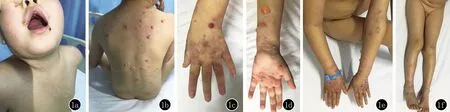
图1 软腭水疱和糜烂面,口周水疱,背部、四肢散在红斑、水疱,尼氏征阴性
实验室检查:血常规、尿常规、大便常规、肝肾功均正常;抗Dsg3抗体、抗Dsg1抗体、抗BP230抗体、抗BP180抗体、抗VII胶原抗体均为阴性。胸部X线片、心电图均正常。腹部B超:肝右叶钙化灶(多发)。光试验:UVA-MED 31.9 J/cm2(参考范围≥20 J/cm2),UVB 15M J/cm2(参考范围 ≥23 MJ/cm2)。组织病理(背部)示:表皮下水疱,真皮乳头中性粒细胞、少许嗜酸粒细胞小脓肿,浅层中性粒细胞、嗜酸粒细胞、淋巴细胞浸润(图2a、2b)。直接免疫荧光:表皮基底膜IgA阳性、C3弱阳性线状沉积,IgG、IgM阴性(图2c、2d)。间接免疫荧光:表皮基底膜循环抗体IgG、IgA 1∶40阳性,IgM、C3阴性。正常人皮肤盐裂间接免疫荧光:循环抗体IgA沉积在表皮侧;IgG沉积在表皮侧,局部区域表皮真皮双侧。
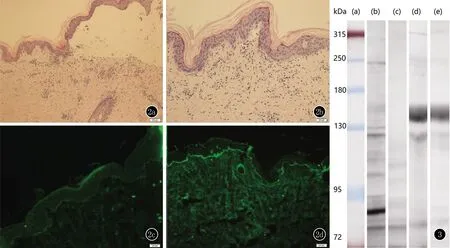
图2 2a:表皮下水疱(HE,×100);2b:真皮乳头嗜中性粒细胞少许嗜酸粒细胞小脓肿,浅层中性粒细胞、嗜酸粒细胞、淋巴细胞浸润(HE,×400);2c:表皮基底膜C3弱阳性线状沉积(DIF,×200);2d:表皮基底膜IgA阳性线状沉积(DIF,×200)
免疫印迹示IgA表皮侧近95 kDa、近130 kDa、130 kDa、250 kDa均有条带(图3)。
诊断:儿童LAGBD。治疗:甲泼尼龙40 mg静滴 每日1次,卤米松乳膏外用,每日2次。至住院第5天原有皮损消退不明显,仍有新发水疱,加用氨苯砜50 mg口服每日1次,于住院第14天增量至75 mg每日1次。患儿仍有新发水疱,遂于住院第17天加用人免疫球蛋白10 g静滴,每日1次,连续应用5天,患儿未再有新发水疱。住院第22天调整糖皮质激素用量为甲泼尼龙32 mg静滴每日1次,住院第23天给予吗替麦考酚酯0.25 g口服,每日3次。随后于住院第25天、28天、31天分别调整糖皮质激素用量为甲泼尼龙24 mg口服,每日1次、甲泼尼龙16 mg口服每日1次、甲泼尼龙8 mg口服每日1次。住院第35天调整氨苯砜用量为100 mg口服,每日1次,增加卤米松/三氯生软膏外用。患儿住院第36天停用糖皮质激素,其余药物用法、用量不变,偶于背部,前臂新发粟粒大水疱,增加外用药涂抹频率后可逐渐消退。治疗51天后,原有皮疹基本消退,无新发水疱,遗留多处色素沉着(图4),好转出院。
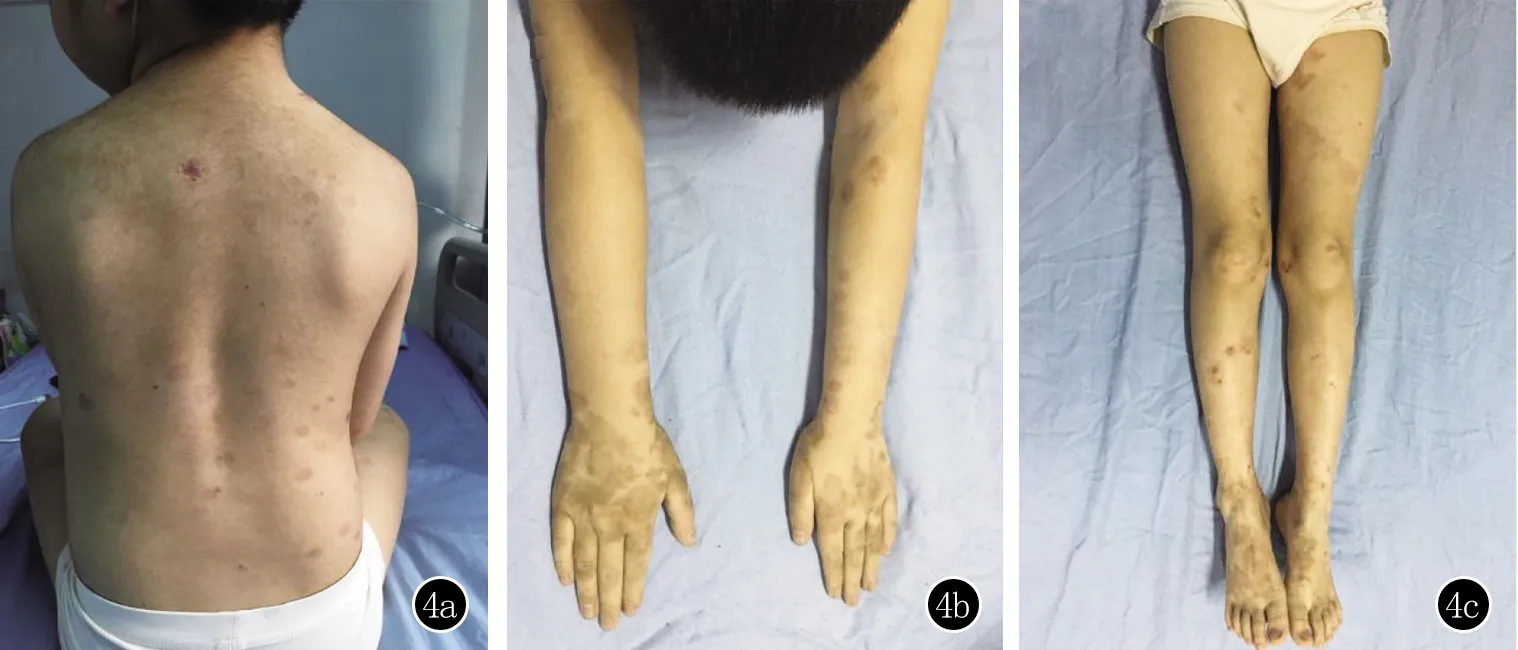
图4 患者出院当天皮肤科查体:背部(4a),双侧前臂(4b)、双下肢(4c)等处原有皮疹基本消退,无新发水疱,遗留色素沉着
患儿出院后第8天下肢远端及眼睑出现米粒大水疱,增加外用药涂抹后消退不明显。遂于出院后第15天加用醋酸泼尼松4片口服 每日1次,皮疹逐渐消退。随后于出院后第35天、51天分别调整醋酸泼尼松用量为3片口服 每日1次,醋酸泼尼松2片口服 每日1次。现患儿皮疹完全消退,一般情况良好,血常规、肝肾功等检查无异常,激素已调整为醋酸泼尼松口服 双日2片,单日1片,吗替麦考酚酯用量调整为0.25 g口服 每日2次,氨苯砜用法、用量不变。患儿目前仍在随访中。
讨论LABD是一种少见的,以基底膜带上连续性线状IgA抗体沉积为特点的自身免疫性疱病,其典型临床表现为腊肠样环形排列的水疱,由Bowen[3]于1901年首次描述,但当时仍被认为是疱疹样皮炎。Jablonska等[1]于1979年提出该病是一种独立疾病,并将其命名为LABD。该病的发病率在不同的国家和地区有所差别,从0.22/106~1.45/106不等,乌干达等非洲国家发病率稍高[4]。据文献报道,该病的发生可能与HLA Cw7、HLA B8、DR3、DQ2等基因有关[4]。部分患者发病前无明显诱因,称之为特发性LABD,而其余患者发病前有明显诱因,如药物、肿瘤、感染、紫外线等[5-8]。LABD的自身抗原主要位于透明层和致密层,根据IgA抗体针对的自身抗原所在的位置不同,分为透明层型和致密层型,透明层型LABD的主要抗原是97-kDa(LABD-97)抗原和120-kDa(LAD-1)抗原,而致密层型LABD的主要抗原是255-kDa真皮抗原和290-kDa(VII胶原)抗原[9-12]。
部分被诊断为LABD的患者血清中同时存在抗基底膜带的IgG抗体,这种情况由Zone等[2]于1994年首次报道,并将其命名为LAGBD,在他们的研究中,IgG抗体针对的抗原具有异质性,包括97-kDa抗原、180-kDa抗原、230-kDa抗原等。后续对LAGBD患者血清中IgG所针对抗原的研究较少,主要是laminin-332、laminin-γ1、integrin α6β4、190-kDa表皮抗原、BP180羧基端抗原、BP180NC16a等[13-15]。LAGBD的临床表现与LABD类似,皮疹主要为躯干、四肢红斑,紧张性环形水疱、大疱,部分患者可累及黏膜,瘙痒明显;直接免疫荧光显示基底膜带上有IgA抗体呈线状沉积的同时伴有IgG抗体沉积,或间接免疫荧光显示抗基底膜带IgA、IgG循环抗体阳性,或正常人皮肤盐裂间接免疫荧光显示IgA、IgG抗体沉积,或通过免疫印迹、ELISA等技术检测出抗基底膜带IgA、IgG循环抗体阳性[1,15-17]。LAGBD的治疗同样与LABD相似,主要是口服氨苯砜,部分效果不佳的患者可联合糖皮质激素,吗替麦考酚酯或者人免疫球蛋白[13,17-21]。
LABD和LAGBD的关系目前尚未清楚,有学者认为两者是同一种疾病[16],还有学者认为LABD和大疱性类天疱疮是一组疾病谱的两端,而LAGBD位于此疾病谱的中间[15]。
本例为男性患儿,既往体健。发病前一天曾于露天泳池长时间“暴晒”,当晚暴露部位皮肤稍红,偶有瘙痒,次日开始起皮疹。追问病史,患儿此前多次日晒后出现暴露部位变红,自行消退慢。否认近期感染史、服药史、肿瘤史等病史。患儿光试验提示对UVB不耐受。综上考虑紫外线诱发可能性大。患儿直接免疫荧光提示表皮基底膜IgA阳性,C3弱阳性线状沉积,间接免疫荧光及正常人皮肤盐裂间接免疫荧光均显示IgA、IgG循环抗体阳性,符合儿童LABD、LAGBD。患儿病情顽固,住院时间长,治疗上除使用甲泼尼龙、氨苯砜外,还联合人免疫球蛋白、吗替麦考酚酯等多种药物,过程复杂。目前国内未见紫外线诱发难治性儿童LAGBD的报道,我们的病例为该类疾病的诊疗提供了一定思路。同时,关于该疾病的进一步诊疗规范及该患者病情顽固性的原因仍需进一步探讨。
猜你喜欢
听力学及言语疾病杂志(2022年5期)2022-09-20 09:07:10
山东冶金(2022年2期)2022-08-08 01:50:44
山东冶金(2019年1期)2019-03-30 01:34:54
振动与冲击(2018年4期)2018-03-05 00:34:24
实用临床医药杂志(2016年21期)2016-12-09 03:28:12
兽医导刊(2016年12期)2016-05-17 03:51:42
兽医导刊(2016年12期)2016-05-17 03:51:36
山东青年(2016年2期)2016-02-28 14:25:33
应用化工(2014年10期)2014-08-16 13:11:29
振动与冲击(2014年23期)2014-05-16 07:01:56
