
Physical exercise may exert its therapeutic in fluence on Alzheimer’s disease through the reversal of mitochondrial dysfunction via SIRT1-FOXO1/3-PINK1-Parkin-mediated mitophagyT
2021-02-03 07:08NaZhaoJieXiaBoXu
Na Zhao,Jie Xia,Bo Xu,*T
aKey Laboratory of Adolescent Health Assessment and Exercise Intervention of Ministry of Education,East China Normal University,Shanghai 200241,China
bCollege of Physical Education and Health,East China Normal University,Shanghai 200241,China T
Received 18 June 2020;revised 26 July 2020;accepted 6 August 2020 Available online 28 August 2020
2095-2546/©2021 Published by Elsevier B.V.on behalf of Shanghai University of Sport.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/)
Alzheimer’s disease(AD)is the most common form of agerelated neurodegenerative disease.Importantly,the prevalence of AD is expected to increase as the population ages,and it will place a huge burden on health care in the future.AD is clinically characterized by confusion,disorientation,and progressive memory and language decline.Diagnosis is con firmed histologically by the accumulation of extracellular β-amyloid(Aβ)plaques,intracellular tangles of abnormally phosphorylated tau protein,and progressive neuronal loss in the brain.1Current treatment focuses on symptom alleviation since there is not yet a cure for AD,despite the wealth of research on the mechanisms underlying the disease and its pathophysiology.2
Interestingly,numerous investigations clearly suggest a bene ficial role of physical exercise in AD.3In fact,it is thought that physical exercise may not only prevent the progression of AD,but also mitigate the risk of developing the disease.Various mechanisms have been proposed for the bene ficial properties of exercise in AD,including a reduction in abnormal protein aggregates and neuroin flammation and a boost in neurogenesis.3However,the underlying mechanisms accounting for the bene fits of exercise are not well-understood.Identifying these mechanisms could help promote the development of novel pharmaceutical agents in the treatment of AD.
Recent studies have highlighted the contribution of mitochondrialdysfunctionandbioenergeticde ficittotheAβ andphosphorylated tau depositions in the brain of AD patients and animals,suggesting that mitochondrial dysfunction is an early event in AD pathology.4In addition to reduced activity of mitochondrial respiratory enzymes,ultrastructural de ficits,including the loss of cristae,the development of vacuolar intramedullary lesions,and increased permeability,have been demonstrated in brain tissue in AD patients.5Consequently,much evidence has been reported showing that excessive reactive oxygen species and reduced adenosine triphosphate(ATP)levels are present in neurons,thereby exacerbating mitochondrial dysfunction and triggering apoptoticneuronalcelldeathinAD.6In fact,multiplelinesofevidence have shown that exercise can play a therapeutic role in the development and progression of AD via promoting mitochondrial f i tness(see the review by Bernardo et al.7).In addition to decreased reactive oxygen species production,other exerciseinduced effects,such as changes in glucose uptake,activity of mitochondrial respiratory enzymes,ATP levels,and mitochondrial biogenesis,have all been described as bene fits of exercise in combatting mitochondrial dysfunction in AD.8-10However,the underlying mechanisms accounting for the bene fits of exercise on mitochondrial fitness are still elusive.
In this opinion piece,we propose a novel idea:physical exercise may exert its therapeutic in fluence on AD through the reversal of mitochondrial dysfunction via a Sirtuin1(SIRT1)-mediated mitophagy.Under physiological conditions,damaged mitochondria are surrounded by autophagosomal vesicles,before fusion with lysosomes.The resulting autolysosomes are crucial in the removal of the damaged mitochondria through the action of proteases and other catabolic enzymes.Interestingly,postmortem brain tissue examination in AD patients exhibits abnormal accumulation of mitochondrial constituentswithin autophagosomalvacuoles.11,12Abnormal changes in the mitophagy-linked pathway PARK6(phosphatase and tensin homologue(PTEN)induced putative kinase 1(PINK1))-Parkin have been found to contribute to AD.13Notably,recent publications14,15inNatureandNature Neurosciencesuggest that impaired mitophagy activity plays a critical role in AD onset and progression.The authors of these 2 studies discovered that nicotinamide adenine dinucleotide(NAD+)precursors,such as nicotinamide mononucleotide and nicotinamide riboside,could inhibit Aβ and phosphorylated tau aggregation,improve mitochondrial dysfunction,and reverse cognitive de ficits in GMC101 worms and APPswe/PS1De9(APP/PS1)mouse models of AD via enhancing PINK1-Parkin-dependent mitophagy activity.14,15Although NAD+replenishment may serve as a promising candidate for combatting AD in animals,how to translate it to humans remains a challenge.
It is common knowledge that NAD+is an essential cofactor for SIRT1.A seminal paper from 2012 suggested that a high NAD+/nicotinamideadeninedinucleotide(reduced)(NADH)ratio can mediate mitophagy through an increase in SIRT1 activation.16Previous studies have revealed that the NAD+-dependent SIRT1 upregulates mitophagy by activating theforkheadbox O1/3(FOXO1/3)-PINK1-Parkinpathway.17,18In this regard,extensive research has con firmed that physical exercise significantly alters the NAD+/NADH ratio and results in enhanced expression of SIRT1 in the brain.19More important,exercise has been shown to attenuate cognitive decline,improve synaptic dysfunction and enhance survival factors,and decrease the Aβ burden via the activation of the SIRT1 signaling pathway in mouse models of AD.20,21
Moreover,it has recently been demonstrated that treadmill exercise activates autolysosomes function in wild-type mice brains through the SIRT1 pathway,and it has also been found that inhibiting the deacetylation effect of SIRT1 abolishes the transcription of lysosomal genes that are regulated by exercise.22Recent studies have demonstrated that PINK1-Parkindependent mitophagy is enhanced after treadmill exercise,resulting not only in increased resistance to age-related and doxorubicin-induced mitochondrial alterations but also in attenuated Aβ-induced cognitive decline and mitochondrial dysfunction in AD animal models.23-25It is worth mentioning that long-term exercise is associated with the improvement of mitochondrial quality control in the hippocampus of aged Sprague Dawley rats and that the autophagy pathway is essential for this process.26Therefore,we suggest that exercise is likely to enhance mitophagy activity by activating the SIRT1-FOXO1/3-PINK1-Parkin pathway through a high NAD+/NADH ratio,thus contributing to the mitigation of the mitochondrial dysfunction associated with AD neurodegeneration.Alternatively,activating the SIRT1 pathway by physical exercise balances both mitophagy and mitochondrial biogenesis,allowing for the maintenance of the functional integrity of mitochondria in the brains of AD(Fig.1).
It is worth mentioning that we cannot exclude the possibility that SIRT1 may exert its therapeutic in fluence on AD through pathways other than the mitophagy pathway.For example, findings suggest that lactate mediates the effects of exercise on learning and memory through SIRT1-dependent activation of hippocampal brain-derived neurotrophic factor(BDNF).27We believe that an exercise-induced increase in BDNF levels is closely related to the activation of SIRT1.A great deal of evidence has con firmed that SIRT1 is an important upstream molecule that activates BDNF.28,29Perhaps,the bene fits of exercise lie in the diversity of “pills of exercise”,and these “pills”perform dual functions by regulating multiple factors,including SIRT1 and BDNF.
We believe that the bene fit of exercise on mitophagy is of broad interest to the neuroscienti fic community and that the results of this interest will inspire further studies that investigate the role of SIRT1-mediated mitophagy in AD prevention and treatment.
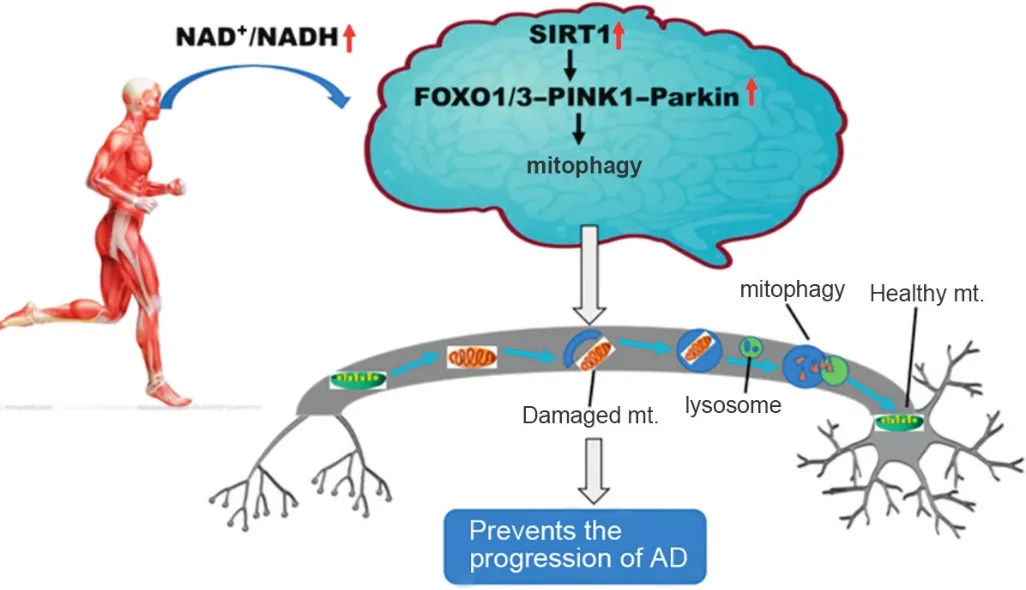
Fig.1.Physical exercise may exert its potential curative effects in Alzheimer’s disease(AD)viaSIRT1-FOXO1/3-PINK1-Parkin-mediatedmitophagy.NAD+/NADH ↑means that the NAD+/NADH ratio is increased;SIRT1↑means that SIRT1’s activity is increased;FOXO1/3-PINK1-Parkin ↑means that FOXO1/3-PINK1-Parkin signaling pathway is up-regulated.FOXO=forkhead box O;mt.=mitochondria;NAD+=nicotinamide adenine dinucleotide;NADH=nicotinamide adenine dinucleotide(reduced);PINK1=phosphatase and tensin homologue(PTEN)induced putative kinase 1;SIRT1=Sirtuin1.
Acknowledgment
This study was supported by the Fundamental Research Funds for the Central Universities(40500-20104-222290).
Authors’contributions
NZ was responsible for the overall concept and content of this opinion article and writing the first draft of the manuscript;JX and BX critically reviewed the manuscript.All authors have read and approved the final version of the manuscript,and agree with the order of presentation of the authors.
Competing interests
The authors declare that they have no competing interests.
 Journal of Sport and Health Science2021年1期
Journal of Sport and Health Science2021年1期
- Journal of Sport and Health Science的其它文章
- Author biographies of Editor-in-Chief’s choice
- Association between physical activity and digestive-system cancer:An updated systematic review and meta-analysisT
- Risk factors for overuse injuries in short-and long-distance running:A systematic reviewT
- Exercise interventions for older adults:A systematic review of meta-analysesT
- he mirror’s curse:Weight perceptions mediate the link between physical activity and life satisfaction among 727,865 teens in 44 countriesT
- Sensor-based physical activity,sedentary time,and reported cell phone screen time:A hierarchy of correlates in youthT