
水菱镁矿制备高分散六角片状纳米氢氧化镁的新工艺条件探究
2021-02-02 11:27黄建翠凌观爽
无机盐工业 2021年2期
黄建翠,凌观爽,宗 俊
(华东师范大学化学与分子工程学院,上海200241)
氢氧化镁因兼具阻燃、无毒、抑烟、环保、热稳定性好及填充安全等优点, 已成为高聚物阻燃领域中极具发展前途的阻燃剂之一[1-4],但常规合成的氢氧化镁颗粒较大,形貌多为无定形或六角薄片,具有极性强、结晶性差、比表面积大、易发生二次团聚的特点,与高分子材料的兼容性较差,从而严重影响了复合材料的力学性能和阻燃性能[5]。纳米氢氧化镁(1~100 nm) 具有纳米材料所具有的共性特点, 如小尺寸效应、量子尺寸效应、表面效应等,将其应用于高聚物中能强化材料的性能。 因此,制备纳米级、高分散、 晶形完整和低比表面积的氢氧化镁是当前国内外研究的重要方向。
通常,产物形貌、颗粒大小、粒径均匀程度和分散性强烈依赖于 Mg(OH)2的制备过程[6]。 迄今为止已制备出多种形态的氢氧化镁晶体,如板状、层状、棒状、针状等。 目前,常用的阻燃型纳米氢氧化镁制备方法主要为湿法共沉淀法[5-8]、水热法[9-11]、氧化镁水化法[12]等。 前人的工作多数围绕不同试剂间的沉淀反应,通过控制不同条件制备氢氧化镁,然而这种方法需要大量的化学试剂,成本高昂,不利于工业化生产。因此,利用中国丰富的矿产资源制备优质镁产品是当前镁化合物生产研究趋势。
水菱镁矿是一种储量丰富的天然碱式碳酸盐矿物。 水菱镁矿质地纯净, 色泽洁白, 其化学式为4MgCO3·Mg(OH)2·4H2O[13],其中 CaCO3等杂质含量低,对一般性应用影响很小,是制备阻燃剂、氧化镁、重质/轻质碱式碳酸镁、氢氧化镁等镁质产品的优质矿产原料[14-15]。 相比菱镁矿、白云石等常用镁矿而言,采用水菱镁矿制备纳米氢氧化镁具有无需除杂、无需酸解、操作简单、环保、性价比高的优势。
本文提出了以水菱镁矿为原料,通过“煅烧-水化-煅烧-水热”的路线制备分散性良好的六角片状纳米氢氧化镁的方法。 很多研究表明,在Mg(OH)2的结晶过程中经过水热与改性可以增加在 (001)极性平面上生长的晶体数量, 从而增加I001/I101和I001/I110的值,进而改善颗粒的分散性和稳定性[5]。 因此,本文继续探究了水热与改性过程中各因素对氢氧化镁结晶度、形貌及分散性的影响。
1 实验部分
1.1 实验原料
天然水菱镁矿[4MgCO3·Mg(OH)2·4H2O]取自西藏班戈湖那曲。 聚乙二醇 6000(PEG6000)、聚乙二醇 200(PEG200)、聚乙烯吡咯烷酮(PVP)、十六烷基三甲基溴化铵(CTAB),均为分析纯。 去离子水实验室自制。 所有试剂无需进一步纯化即可使用。
1.2 纳米氢氧化镁制备方法
1.2.1 煅烧
将适量水菱镁矿石放入坩埚中,一同放入马弗炉中煅烧。 设置煅烧温度为650 ℃、煅烧时间为30 min、升温速率为 10 ℃/min。 冷却到室温后,将其研磨至粒径≤75 μm,得到白色粉末,记为M1。
1.2.2 水化
将去离子水加入到三颈烧瓶中并加热至80 ℃,称取一定量的上步制备的M1,在剧烈搅拌下缓慢加入到三颈烧瓶中,保持固液比为1∶30(质量比),水浴回流反应2 h。反应结束后,悬浊液陈化30 min,抽滤,并用去离子水洗涤滤饼3 次。 将滤饼放置在150 ℃鼓风干燥箱中烘干3 h,研磨得白色粉末,记为MH1。
1.2.3 煅烧
将MH1置于坩埚中再放入马弗炉中煅烧,设置煅烧温度为650 ℃、煅烧时间为60 min、升温速率为10 ℃/min。 冷却至室温,所得白色粉末记为 M2。
1.2.4 水热
分别配制不同质量分数(1%~30%)的M2与去离子水混合物,放入聚四氟乙烯高压反应釜中,分别在不同温度(100、150、200 ℃)下反应一段时间(6、3、1、0.5 h)。 反应结束后,所得悬浊液自然冷却至室温(20 ℃),将产物进行真空抽滤,所得滤饼用去离子水洗涤3 次。 将滤饼放置在110 ℃鼓风干燥箱中烘干12 h,研磨得白色粉末,记为MH2。
1.2.5 改性
将用量为氧化镁质量4%的不同改性剂(PEG200、PEG6000、PVP、CTAB)分别与去离子水混合均匀,按1.2.4 节做水热反应,探究不同改性剂对六角片状纳米氢氧化镁的分散性的影响。而后改变改性剂用量,探究不同用量(2%、4%、6%,质量分数,下同)的改性剂对六角片状纳米氢氧化镁分散性的影响。
1.3 表征方法
用SmartLab SE 型X 射线粉末衍射仪做物相分析,参数设置:Cu 靶 Kα 辐射,λ=0.154 18 nm,镍滤光片,25 mA,35 kV,扫速为 30(°)/min;用 TGA/SDTA851e 型差热热重分析仪分析水菱镁矿的热分解过程,参数设置:温度范围为室温~800 ℃,升温速率为 10 ℃/min,N2气氛;用 Quanta FEG 250 型场发射扫描电子显微镜观察产物形貌。
2 结果与讨论
2.1 水菱镁矿原料的表征
图1a 为天然水菱镁矿的XRD 谱图。由图1a 可见,主要衍射峰与 4MgCO3·Mg(OH)2·4H2O(JCPDS 70-0361)和 CaCO3(JCPDS 41-1475)的峰 位置 几乎一致,证明主要矿物组成是水菱镁矿[4MgCO3·Mg(OH)2·4H2O]和文石(CaCO3)。 样品经化学分析,测得其中氧化镁质量分数为41.59%,氧化钙质量分数为1.30%,其他杂质含量较少。
水菱镁矿在800 ℃氮气气氛中热分解, 得到TG-DTG 曲线,见图 1 b。 由图 1b 的 TG 曲线可见,样品的初始分解温度为150 ℃左右, 分解完全的温度在680 ℃附近。 根据TG 曲线的质量损失台阶可将样品的质量损失分解过程分为4 个区间:150~350 ℃、350~480 ℃、480~560 ℃、560~680 ℃。 由图 1b 的 DTG曲线可以看出,各阶段的质量损失率不同,4 个温度区间的质量损失分别为 13.33%、29.28%、8.28%和3.51%,总的分解率为54.40%,其总吸热反应分解的方程式见式(1),因此,采用650 ℃煅烧基本可以完全分解。
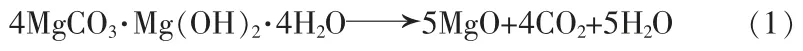
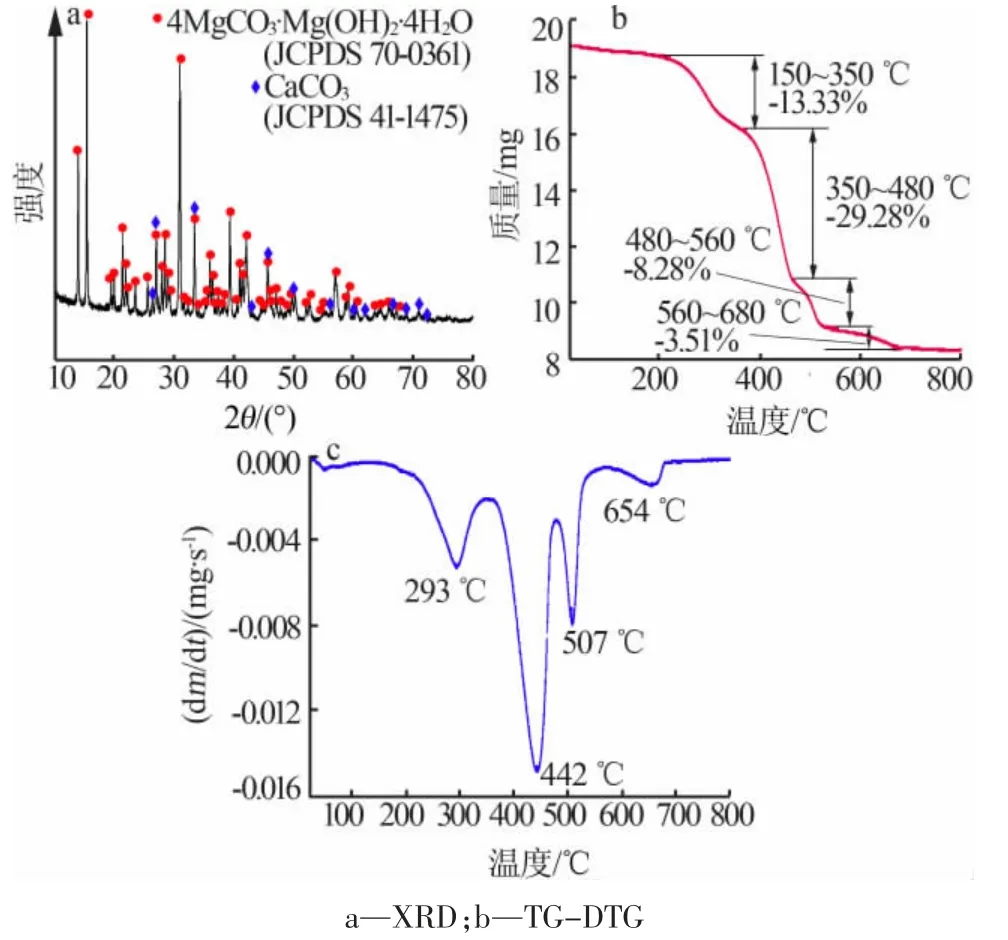
图1 天然水菱镁矿XRD 与TG-DTG 谱图
2.2 不同方法制备纳米氢氧化镁的表征
在确定以水菱镁矿为原料制备六角片状纳米氢氧化镁的上述制备方法之前, 为寻找最佳的工艺路线,分别研究了a 路线“煅烧-水化-煅烧-水化”、b路线“煅烧-水化-水热”、c 路线“煅烧-水热”、d 路线 “煅烧-水化-煅烧-水热”[每一步的制备方法与上面所介绍的制备方法一致, 其中水热步骤中使用的氧化镁用量为5%(质量分数)、 温度为150 ℃、时间为6 h], 研究4 种不同的制备路线对纳米氢氧化镁形貌的影响,并通过XRD、SEM 技术做了表征。
图2A 为不同方法路线制备的样品的XRD 谱图。 由图 2A 可见, 衍射峰主要位于 18.58、32.84、37.96、50.88、58.70、62.14、68.10、72.16°处,与标准卡片 JCPDS 44-1482 中 Mg(OH)2的峰基本一致,证明了制备的产物均为Mg(OH)2。通过4 种方法的XRD谱图比较, 可以看出d 路线 “煅烧-水化-煅烧-水热”方法所制备的氢氧化镁的峰强度明显增高,峰形也更加尖锐。 图2A 显示在d 路线下,各个晶面的半峰宽最窄,平均晶粒尺寸最大,且I001与I101、I110的衍射峰强度之比最大。(001)方向的表面能低于所有其他方向[例如(101)和(110)]的表面能[21],也就是说(001)方向的极性比(101)方向的极性弱。 晶体在(001)晶面上生长,这可能导致 Mg(OH)2颗粒的内部应力和表面极性降低, 从而能提供更大的结构稳定性,减少颗粒的团聚,改善颗粒的分散性能。 以上结果表明,相比 a、b、c 路线,在 d 路线下,氢氧化镁的结晶度最好, 晶形也更完整。 通过对比图2B 的SEM 照片可以看出,a 路线下,样品中仅有少量较规则的六角片纳米氢氧化镁,大部分晶形不完整。b 路线下, 样品显示为各种不规则的薄片杂乱无章地团聚在一起。 c 路线下,样品的片状模糊,且未形成规则的六角片状,团聚现象非常严重。 而在d 路线下,样品的六角片状非常规整,棱角清晰,且层与层之间分隔明显,片的厚度为40~60 nm,这与XRD 谱图的结论相吻合。 因此,选择d 路线即“煅烧-水化-煅烧-水热”的工艺路线来制备六角片状纳米氢氧化镁。
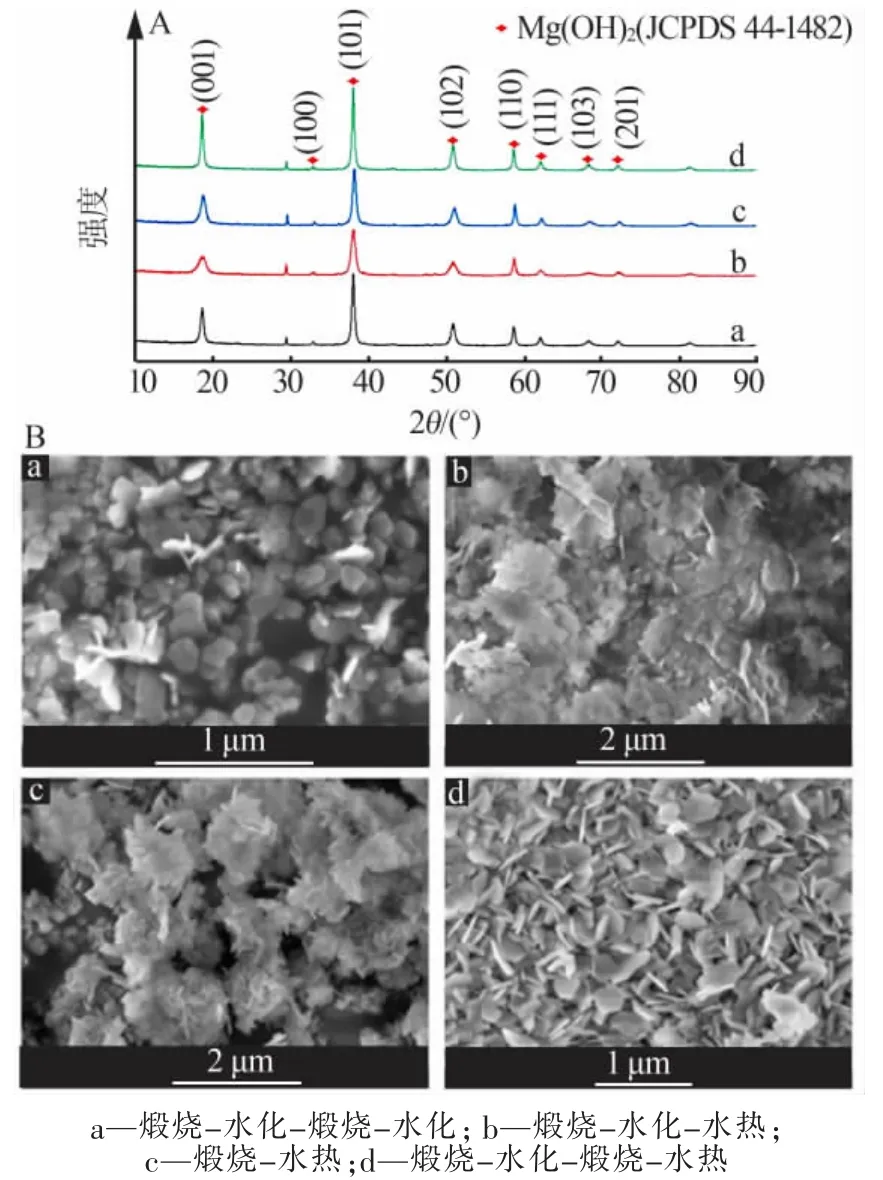
图2 不同方法制备纳米氢氧化镁的XRD 谱图(A)和 SEM 照片(B)
2.3 d 路线中过程产物的表征
图3 为水菱镁矿在水热前每步反应所得产物的XRD 谱图。 从图3 二次煅烧后的XRD 谱图可以看出,煅烧产物的衍射峰主要位于36.76、42.84、62.12、74.64、78.14°处,与 MgO(JCPDS 89-4248)的峰完全符合,说明煅烧产物为MgO。 水化产物的衍射峰主要位于 18.58、32.84、37.96、50.88、58.70、62.14、68.10、72.16°处,与 Mg(OH)2(JCPDS 44-1482)的峰完全吻合,说明水化产物是 Mg(OH)2。 另外,还存在一些杂质CaCO3。
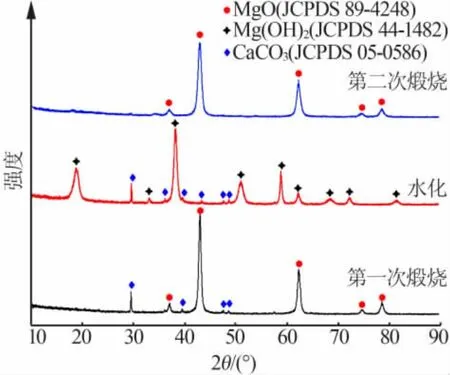
图3 过程产物的XRD 谱图
实验对水菱镁矿在水热前每步所得产物做了SEM 测试(本文SEM 照片均可扫描文章首页OSID码查看)。 结果发现,水菱镁矿原料经首次煅烧后得到的氧化镁M1无特定形貌; 经水化的MH1形貌呈薄片状,片与片之间交叉生长,形成“卡房结构”;二次煅烧后得到的氧化镁M2在形貌上保持了前驱体的结构,具有多孔性,呈现“网”状结构。
以上过程步骤中虽然发生了形貌的改变, 但并未得到明显六角片状的氢氧化镁,因此“水热”是制备六角片状纳米氢氧化镁的关键步骤。 接下来解释了水热过程可能的反应机理, 并采用单因素控制变量法考察了水热与改性过程中各因素对六角片状纳米氢氧化镁形貌及分散性的影响, 进而得出最佳水热改性条件。
2.4 水热反应机理
水热反应可以很好地改善颗粒的结晶度和分散性。 这可能归因于在水热体系中遵循着 “溶解-沉淀-溶解-重结晶”的反应机理,可以促进晶体的发育生长,发生的主要化学反应和反应机理如下。
1)溶解。 MgO 在水中提供电子,生成 OH-,OH-吸附在带正电荷的固体表面,OH-从表面进入溶液中,同时释放出Mg2+。
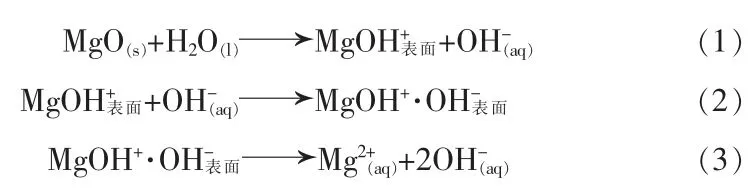
2)沉淀。离子浓度增大形成过饱和溶液,此时氢氧化物在氧化物表面生成沉淀。
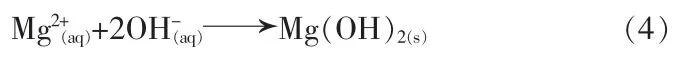
3)溶解。 生成的 Mg(OH)2晶体在高温、高压下再一次溶解为可溶性 Mg(OH)2,可溶性 Mg(OH)2转变为 MgOH+和 OH-,MgOH+继续溶解为 Mg2+和 OH-。
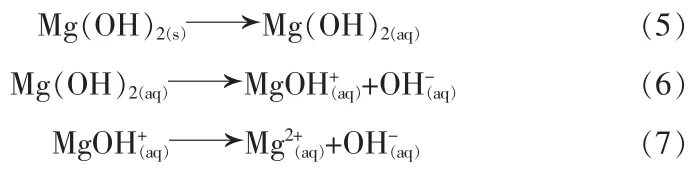
4)重结晶。 再次溶解的Mg2+和OH-形成过饱和溶液,沉淀再次形成。
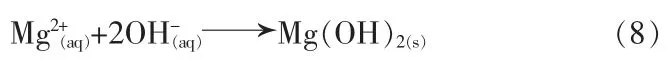
2.5 最佳水热条件探究
2.5.1 MgO 用量
图4 为不同质量分数的M2进行水热实验的XRD 谱图。 由图 4 可以看出, 随着 MgO 用量的增加,Mg(OH)2的特征峰峰强总体呈先增大后减小的趋势, 而半峰宽先减小后增大,I001与 I101、I110的衍射峰强度之比先增大后减小(XRD 补充信息可扫描文章首页OSID 二维码查看)。 表明水热过程中,MgO用量对晶体的生长方向产生了一定的影响。 通过SEM 观察可以看出,M2的形貌均为规则的六角片状,且随着MgO 用量的增加,六角片厚度先增加后减小,在用量为10%~25%(质量分数)时厚度大小较均匀,平均厚度为78 nm 左右。 因此,实验选择适宜的MgO 添加量为10%~25%。
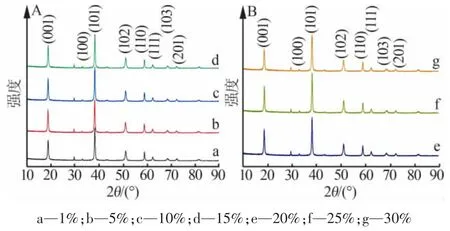
图4 由不同质量分数M2 制备的MH2 的XRD 谱图
2.5.2 水热温度及水热时间
图5A 为不同水热温度下反应所得MH2的XRD谱图。 由图 5A 可以看到,当温度从 100 ℃升至150 ℃,氢氧化镁衍射峰的峰强度随之增大,峰型更加尖锐,表明升高温度有利于晶形发育。 峰强比I001/I101从0.507 增大为0.647,可归因于较高的温度有利于(001)晶面方向生长以及可获得较高的溶解和运输速率[16]。 继续增大温度至 200 ℃,峰强度减弱,半峰宽值增大,说明温度过高反而不利于晶形发育。通过SEM 测试可知,100 ℃水热处理后的产物形貌呈现出较薄的六角片状,颗粒之间团聚明显。当水热温度为150 ℃时,六角片状片层厚度增加,分散性有所改善。控制温度在200 ℃时,氢氧化镁六角片状数量减少,并且出现许多小颗粒晶体,片层厚度减薄。因此升高温度,能促进极性弱的(001)晶面生长,改善形貌和分散性;但温度过高,会导致一部分晶体溶解。这与XRD 表征得到的结论一致。因此,确定最佳水热温度为150 ℃。
图5B 为不同水热时间获得的MH2样品的XRD谱图。 从图5B 可以看出,随着水热时间从0.5 h 延长至6 h,氢氧化镁各衍射峰的峰强明显增强。 分析数据表明,相比于(101)晶面和(110)晶面,氢氧化镁(001)晶面的衍射峰强度增长幅度较大,表明延长水热时间对(001)晶面的生长影响较大。 此外,I001/I101的比值和平均粒径的增加以及半峰宽的减小也表明, 延长水热时间有利于氢氧化镁结晶度的提高。

图5 不同水热温度(A)不同水热时间(B)下制备的MH2 的XRD 谱图
通过SEM 观察可以看出,在不同水热时间下均能观察到较规则六角片状结构的氢氧化镁纳米颗粒。 当水热反应0.5~1 h 时, 颗粒之间团聚比较严重;反应3 h 后,呈现出表面光滑、完美的正六角片状结构,厚度为40~60 nm,颗粒团聚有所改善;反应6 h,虽然晶体厚度有所增加,但表面变得粗糙,有些边、角发生溶解,不规则六角片开始增多。 同时考虑到能源消耗问题,实验选择3 h 为最佳水热时间。
2.6 最佳改性条件探究
2.6.1 改性剂种类
向水热反应体系中,分别添加相当于MgO 质量的 4%的 CTAB、PEG6000、PEG200、PVP,在 150 ℃下反应 3 h 得到样品。 图 6 为样品 XRD 谱图。 由图 6 可见,改性前后Mg(OH)2的衍射峰位置保持不变,表明Mg(OH)2产物不受影响。 相比未改性及其他表面活性剂,在PVP 作用下氢氧化镁的I001/I101和I001/I110的值较大。
通过SEM 观察可以看出,加入CTAB、PEG200,其分散性依然较差,颗粒间团聚明显。 相比之下,加入PEG6000、PVP 明显提高了纳米氢氧化镁的分散性,颗粒大小分布均匀,生长方向趋于一致,晶体黏连现象消失。其中,添加PEG6000 时,虽然分散性得以改善,但六角片已经有部分溶解,得到的六角片状已经不规则。 而添加PVP 时,制备的纳米氢氧化镁样品形貌良好, 显现出整齐规则的六边形和均匀的粒径。 这是由于PVP 具有长链结构,能产生空间位阻效应。此外,PVP 分子中的氮原子和氧原子含有孤对电子,能与Mg2+配位,使添加PVP 制得的氢氧化镁晶体具有更加规整的形貌[16]。 因此,选用 PVP 为最佳改性剂。
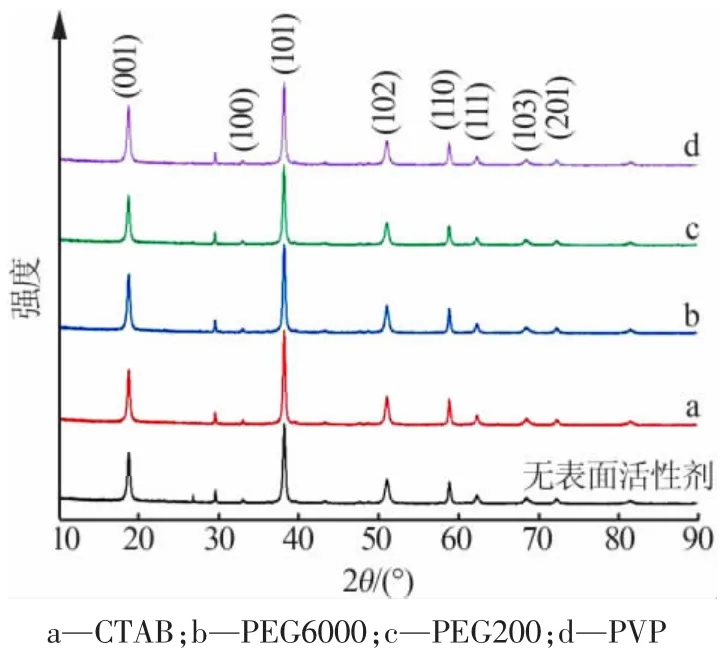
图6 不同改性剂下制备的MH2 的XRD 谱图
2.6.2 改性剂用量
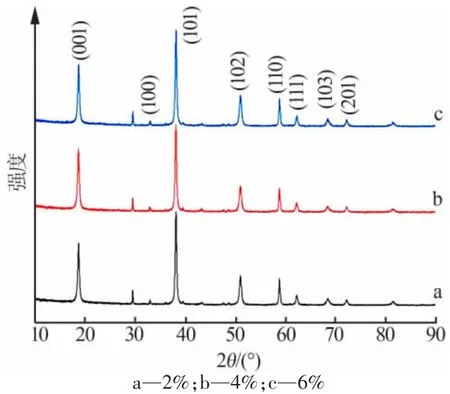
图7 不同质量分数PVP 下制备的MH2 的XRD 谱图
在最佳改性剂PVP 条件下,探究不同质量分数的PVP 对产物分散性的影响。 图7 为不同质量分数PVP 下制备的 MH2的 XRD 谱图。 由图 7 可以看出,随着PVP 的用量的增加,I001/I101和I001/I110的值先增大后减小。
图8 为不同质量分数PVP 下制备的 MH2的SEM 照片。由图8 可以发现,当PVP 用量由2%增至4%时, 六角片的生长方向由较为杂乱变为基本一致,分散性明显提高;然而当用量增至6%时,尽管生长方向趋于一致,但片与片之间堆叠严重,且六角片有所溶解,不规则六角片增多。 因此确定PVP 的最佳用量为4%。

图8 不同质量分数PVP 下制备的MH2 的SEM 照片
3 结论
1)以水菱镁矿为原料,采用“煅烧-水化-煅烧-水热”的简单工艺路线制备出了具有规则六角片、高分散的纳米氢氧化镁。2)实验探究了水热条件,结果表明,在水热过程中,控制水热条件可改变晶体的生长方向,使极性弱的(001)晶面暴露增多,极性大的(101) 晶面暴露减少。 实验最终确定了最佳水热条件:氧化镁用量为10%~25%(质量分数)、水热温度为150 ℃、水热时间为3 h。3)实验探究了改性条件,结果表明,在质量分数为4%PVP 的条件下,氢氧化镁的生长方向趋于一致,分散性得到有效改善,最终得到了具有规整形貌、粒径均匀、平均直径为300~400 nm、六角片厚度为40~60 nm、分散性良好的纳米氢氧化镁。 这归因于PVP 具有长链结构,能产生空间位阻效应,且PVP 分子中的氮原子和氧原子含有孤对电子,能与Mg2+配位,从而使得氢氧化镁晶体具有更加规整的形貌。
猜你喜欢
湖南有色金属(2022年4期)2022-09-17
化工进展(2022年2期)2022-03-09
矿产综合利用(2021年2期)2021-05-24
航空发动机(2021年1期)2021-05-22
建材发展导向(2021年24期)2021-02-12
辽宁化工(2020年12期)2021-01-06
——基于CNKI相关文献的统计分析
金属矿山(2018年9期)2018-10-10
中国塑料(2016年4期)2016-06-27
当代化工(2015年9期)2015-07-10
橡胶工业(2015年8期)2015-02-23
