
加氢裂化反应动力学模型研究及应用进展
2021-02-02 11:27闫乃锋胡智中
无机盐工业 2021年2期
闫乃锋,胡智中
(1.中海油惠州石化有限公司,广东惠州516086;2.中海油天津化工研究设计院有限公司)
随着原油劣质化、重质化趋势的加剧,具备较强原料适应性的二次加工工艺-加氢裂化逐渐受到国内炼厂的青睐[1]。与此同时,加氢裂化兼顾极强的产品灵活性,能够满足不同炼厂需求,在油品加工中的地位显著提升[2]。近年来,加氢裂化装置总体和单个加工规模均明显增加, 中国规模最大的加氢裂化装置(恒力石化)处理量已达380 万t/a。
在加氢裂化工艺逐渐成熟的背景下, 为更好地满足未来原油劣质化和产品苛刻化的需求, 深入了解加氢裂化反应动力学不可或缺[3]。 加氢裂化反应动力学模型能够将加氢裂化原料、产品、催化剂、反应器和工艺条件数字化,再通过模拟计算得出整个工艺过程需求数据, 具有预测快速而精准的优点。模型的应用既可为加氢裂化工艺研究和工业化节省成本,又可支撑企业的智慧炼化建设[4],推进工业信息化进程[5]。因此,加氢裂化反应动力学模型的研究及应用受到了广泛的关注[6]。
本文介绍了国内外加氢裂化反应动力学模型研究历程及最新的研究进展,同时对一些模型的应用情况做了总结,希望能够为加氢裂化反应动力学研究及模型建立提供借鉴。
1 加氢裂化反应动力学模型研究进展
加氢裂化反应动力学模型一直以来都是研究热点,在加氢裂化领域工作者共同努力下,加氢裂化动力学模型已经从起初十分简单的关联动力学模型向复杂的集总动力学模型发展。 其中集总动力学模型衍生出众多分支,分为传统型、连续型和分子型3 种。
1.1 关联动力学模型
关联动力学模型将极其复杂的加氢裂化反应简化为关联公式,代表性的关联式模型有两种。 一种是利用实验室或工业装置的原料、工艺参数等大量数据, 依据一定的计算方式进行收敛回归运算,最终得到产品收率、性质与原料、催化剂、工艺参数等的关联计算公式。 上述动力学模型的研究主要集中在20 世纪60 年代, 代表性研究人员有W.H.Wiser 等[7];另一种关联模型将加氢裂化化学反应与物理规律进行类比,将动力学模型转化为类比模型进行运用,典型代表有正态分布模型和误差函数模型[8]。
加氢裂化关联模型的建立和应用较为简单,在一定时间范围内,当装置、催化剂及工艺条件固定的情况下具有良好的预测性,但缺乏对内部反应规律的描述,在催化剂失活和装置老化等条件下模型极易失效,应用范围很小,基本不具备外推性。 因此,在后续加氢裂化动力学模型研究中关联模型仅供参考,实际研究意义不大。
1.2 集总动力学模型
集总动力学模型按一定的分类方法将反应中的分子划分为不同虚拟组分,常见的分类依据有馏程、化学结构、碳原子数等。 按分类依据可归结为传统集总动力学模型、连续集总动力学模型和分子集总动力学模型 3 类[9]。
1.2.1 传统集总动力学模型
传统集总动力学模型将原料、 中间物料及产物按馏程或其他生产方案需求进行集总划分。当前,该类动力学模型实用性较强、应用广泛。
1939 年 ,S.A.Qader 等[10]建 立 了 二 集 总 模 型 ,按馏程将整个反应体系划分为>350 ℃的原料和<350 ℃的产物两个集总, 该模型拟合度极高且易操作,但原料适应性和产品灵活性太差,难以应用于指导工业实践。1989 年,S.M.Yui[11]提出了三集总模型,建立了原料减压柴油(VGO)转化为轻VGO 和石脑油的反应体系,将氢分压和空速因素键入模型。由于该模型假设加氢裂化反应均为一级反应且反应器恒温,导致该模型的准确性有限。 此后,随着加氢裂化原料、产品及工艺的多样化发展,模型的集总数量逐渐增加,预测的准确性得到了大幅提升。几种典型的不同集总数的动力学模型介绍见表1[12-15],其中R.Krishima 等[15]建立的七集总模型反应网络见图 1。

表1 传统集总动力学模型对比
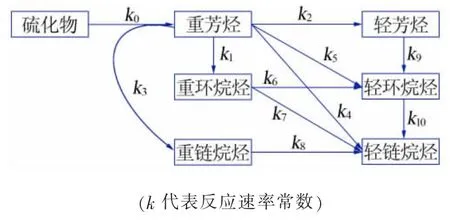
图1 Krishima 七集总模型反应网络
总之, 传统集总动力学模型多以目标产物为基准进行集总划分, 在集总数量较少的情况下准确性有限,增加集总数量可提高模型的准确性,但削弱了模型实用性。 当前针对于固定的加氢裂化装置和工艺, 传统集总动力学模型具备一定的建立可行性和预测效果,是企业建立反应动力学模型的首选。
1.2.2 连续集总动力学模型
连续集总动力学模型的开发基于连续集总理论,选定特性参数实沸点温度馏程(TBP),并结合反应物物化性质和反应选择性, 在特定进料的加氢裂化反应中, 得出混合物蒸馏曲线在反应器中连续变化的趋势,进而对产品收率及性质进行推导预测。
连续集总的概念最早由 De Donder 等提出[16],后 来 C.S.Laxminarasimhan 等[17]依据 连 续 混 合 物 的假设解释了加氢裂化的反应化学过程, 并推导出连续集总模型动力学方程式(1)。若将传统集总模型中各馏分无限细化,其结果与连续集总模型基本相同,说明二者有一定的相似度。 但连续集总模型省略了集总划分步骤,仅对TBP 曲线(油品蒸馏温度与馏出量关系曲线)进行模拟计算分析,需要模型参数更多,模型建立更为复杂。

式中,c(K,t)为 K 组分在反应物料中瞬时浓度,mol/L;p(k,K)为生成 K 组分的理想产率分布函数;D(K)为区分相;dc(K,t)/dt 为 K 组分浓度变化率,mol/(L·s);为生成 K 组分不同速率总和,mol/s;-kc(K,t)为 K 组分裂化速率,mol/s;。
近年来,众多学者基于文献[17]已有建模方法开展了连续集总动力学模型的研究。 P.J.Becker等[18]建立了五组分连续集总模型,将原料替换为链烷烃(P)、环烷烃(N)、不含硫氮芳烃(A)、含氮不含硫芳烃(AN)和含硫不含氮芳烃(AS),并依据TBP馏程分布模拟5 种反应物在加氢裂化过程中的连续反应过程,在模型中反应通过可观的反应路径体现,如图2 所示。该模型将氢化、脱硫、脱氮、裂化等反应用动力学速率方程式表达, 共调用46 个模型参数,模型经过44 个实验测试,单一原料的预测性较优,但原料适应性很差。
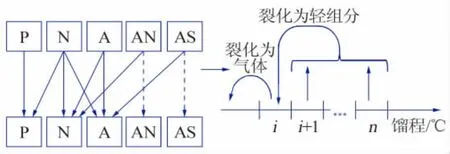
图2 Becker 五组分连续集总模型反应网络
S.S.H.Boosari 等[19]采用改进的连续集总动力学方法模拟加氢裂化过程, 考虑反应器温度对产物分布的影响,对模型做了修正。在4 个不同的反应器温度下, 通过模型最大化预测值和测量数据之间的似然函数来校准模型。 贝叶斯参数估计方法用于关联测量误差和模型预测误差二者的不确定性来获得模型参数的置信区间。 再将蒙特卡罗模拟应用于模型参数的后验范围, 以获得模型中每个单独馏分输出的95%置信区间,在校准模型的输出和测量数据点之间观察到良好的一致性。
连续集总动力学模型能够整合独立的集总,构建连续的反应网络,更加符合实际的反应过程,具备较高的应用价值。 而连续集总动力学模型对参数要求过高,增加了模型建立难度,模型参数的完整性对其预测性影响很大,该模型仍有很大的研究空间。
1.2.3 分子集总动力学模型
近年来,由于“分子炼油”概念兴起,为详尽分析反应器中单一分子的反应历程及速率变化, 通过分子划分集总的动力学模型研究成为当下的热点。 目前, 基于这方面的加氢裂化反应动力学模型有结构导向、单事件和分子管理模型。
结构导向模型建立于分子水平上, 通过对反应物中分子结构进行集总划分, 并结合现有技术获取大量数据信息,从而实现对反应过程定性、定量的预测。D.K.Liguras 等[20]运用基团贡献法,参量选用纯化合物的数据,用向量代表分子反应,计算机程序生成反应网络,最终建立起完整的加氢裂化反应网络,预测反应历程和产物收率;G.G.Martens 等[21]建立的模型结合正碳离子化学理论将集总按碳原子类型划分,参照模型化合物加氢裂化数据,并基于烃类、正碳离子和热力学系数计算集总系数, 较好地预测了工艺条件变化对产品组成的影响;S.Y.Hu 等[22]在文献[21]的基础上从更微观的层次优化动力学模型,通过矩阵变换的计算方式优化了反应器参数, 并实现了对产品的预测;W.J.Song 等[23]建立的模型通过收集工业运行装置数据, 将物料中不同的分子结构与其自创的2D 图形软件进行映射识别, 分子结构通过数字代码表示,最后利用Aspen HYSYS 软件模拟加工过程,其预测结果与实际产品结果相近,该模型建立及运行流程如图3 所示。

图3 结构导向集总模型建立及运行流程
单事件集总模型主要针对基元反应, 每个单一分子作为一个集总。 G.F.Froment[24]应用该方法建立了对同一结构烃类的加氢裂化反应动力学模型,展现了原料组成和中间物料的反应路径, 同时描述了不同烃类化合物的加氢异构化和裂化的反应历程。单事件集总模型在研究机理方面具有较大优势,但其实际应用价值有待挖掘。
分子集总模型在分子层面上管理石油加工过程,其核心是从分子水平认识并优化炼油工艺过程,进而实现油品的高效利用。 史权等[25]对“分子管理”动力学模型做了详尽研究, 该模型建立在对石油分子组成精准定性和精确定量分析基础上; 其次通过特定构造法则对石油分子进行分子构建, 赋予分子特定化学意义,使计算机能够识别各种石油分子,形成石油分子数据库;再通过一定的数据处理方法(例如 D.M.Campbell 等[26]提出的 ARM 方法,或者应用较广泛的蒙特卡洛方法[27]等)对分子反应过程网络进行构建并计算, 最终得出精确的加氢裂化产品收率及性质。该模型建立的反应网络如图4 所示,该网络可将复杂的加氢裂化反应可视化。
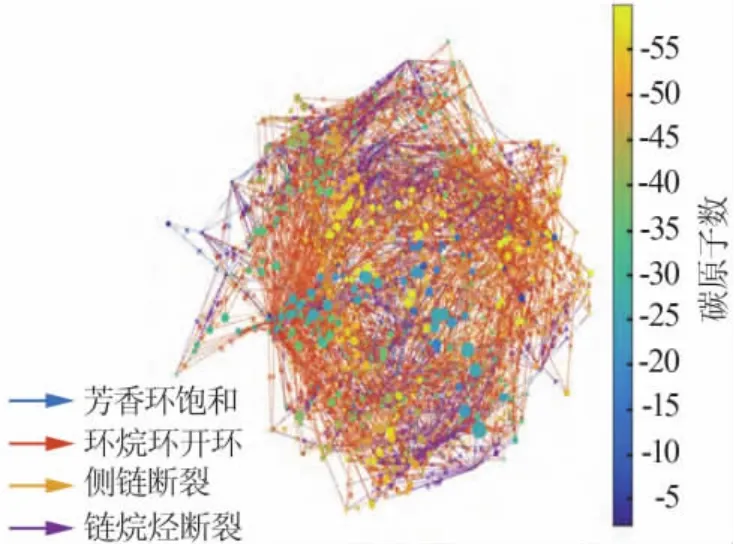
图4 分子集总模型建立的加氢裂化反应网络
分子集总动力学模型基于精确的分析和明确的反应机理, 其发展得益于分析技术的发展和不同工艺反应机理研究的深入, 加之计算机运行速度的加快,模型的应用也能够成为现实,因此分子集总动力学模型已进入快速发展的时代。另外,分子集总动力学模型在分子水平上认识石油组分和石油加工反应过程, 通过对加氢裂化和其他石油加工工艺的优化实现油品的高效利用,应用前景广阔。
2 加氢裂化反应动力学模型应用
对于加氢裂化反应动力学模型来说, 无论是通过关联或集总方法进行构建, 其目的多数是预测产品收率及性质,进而节约成本,为企业创收。因此,精确的预测性是鉴定加氢裂化反应动力学模型优劣的决定性指标。另外,在石油加工过程中,原料、催化剂和工艺条件均可能会发生改变, 因此模型的适用性也是考察其是否具备应用价值的重要指标。
2.1 关联动力学模型应用
R.Krishima 等[15]建立的正态分布关联模型(轴向扩散关联模型)应用十分广泛,该模型将原料和产物通过TBP 曲线关联, 无论反应条件如何变化,产物均有相同的归一化TBP 曲线,预测产品分布平均标准偏差小于10%。 将该模型应用于中试和工业化装置,在低转化率下液体产品分布拟合度较好,但无法预测氢耗、气体产品收率及分布情况。
K.Sharifi 等[28]基于某炼厂加氢裂化装置运行数据建立了一种在线关联动力学模型, 依据映射法则和ASPEN 软件,将装置实时的原料、催化剂、管道、工艺参数等数据均输入程序中, 即可输出产品收率及性质。 模型应用结果表明,装置运行前期10%和末期20%时间段预测效果较差, 结果误差为8%~15%,在运行中期预测结果较好,结果误差在8%以内。 该模型应用效果尚可,但基本不具备外推性。
关联动力学模型的建立和应用十分依赖于中试或工业装置运行数据, 需要从大数据中寻找产物与原料、催化剂、装置、工艺条件等的关联,其外推性较小,应用前景有限。
2.2 集总动力学模型应用
加氢裂化集总动力学模型发展至今建立的模型颇多, 其中连续集总动力学模型受需求参数精确性及操作参数的复杂性限制,仅限于实验室研究阶段,尚未见应用的报道。
2.2.1 传统集总动力学模型
喻胜飞等[29]根据800 kt/a 的蜡油加氢裂化装置运行需求, 以馏程划分建立了四集总模型, 利用高斯-牛顿法对模型参数进行估计,并通过四阶龙格-库塔法计算常微分方程, 实际运行数据表明该模型预测误差小于5%,具有较高的预测精准性。
S.Sadighi 等[12]在建立四集总模型的同时考虑了氢耗因素,其预测精准度提高1.4%,该模型建立时尽可能多地考虑了影响因素,优化了模型。 S.Sadighi等[30]在上述模型基础上,针对 ISOMAX 加氢裂化装置建立了八集总模型,模型增加了新鲜物料流速、反应温度、物料循环速率和催化剂寿命影响参数,其预测结果与实际测定值基本吻合(见图5),预测性较好。企业根据该模型对装置操作条件做了优化,装置利润增加8.17%,经济效益提升显著。

图5 ISOMAX 加氢裂化模型预测结果与实际测定结果对比
郑明方等[14]建立了一种复合型的六集总模型,纵向上集总按馏程划分, 针对不同集总横向上又对其烃类组成按链烷烃、环烷烃、芳烃进行分类,结合25 个模型参数, 根据已有的静态和动态仿真模型,采用正交配置法计算得出最终模型。 将该模型应用于80 Mt/a 加氢裂化装置, 其静态和动态模型预测误差均在5%以下,动态模型预测性能更优。 但该模型未考虑换热及反应温度影响,仍有改进空间。
2.2.2 分子集总动力学模型
Profimatics 公司开发的加氢裂化结构导向集总动力学模型是较为成功的应用模型之一[31],该模型按不同结构烃分子的碳数进行集总划分,烃分子等均按碳数差异归类成不同的虚拟组分, 在分割一系列加氢裂化反应后对反应动力学规律做一定的假设, 最终使模型中各反应和分子基本排除相互影响的可能。 Profimatics 公司依据该模型开发了一整套商品化的加氢工艺软件,具体应用效果未见报道,但从其商业化的结果来看, 该模型具备较高的应用价值。
中石化抚顺石油化工研究院(简称FRIPP)在Powell 的动力学建模理论基础上进一步细化集总,将芳烃、 环烷烃、 链烷烃按化学反应规律有机地结合,再分解形成不同的集总虚拟组分,并严格控制反应入出口总碳数的守恒, 同时排除了反应热力学的影响,并优化了计算方法,采用正算和反算的方法提高模型计算精度。FRIPP 的模型中试应用结果表明,其实验数据与计算数据误差不超过5%。 对于不同原料油和产品切割方案, 该模型均能保证优良的预测性能,是中国加氢裂化集总动力学模型的典范,市场应用空间较大[32]。
Z.Y.Chen 等[33]基于“分子管理”理念,建立了重油加氢过程的仿真模拟动力学模型。 该模型包括2 000 个分子集总,涵盖29 种可能的加氢裂化反应,将反应和分子集总数据输入模型中计算后, 整个反应体系中总计6 000 多个分子和20 000 多个化学反应。将该仿真动力学模型应用于中试实验,其预测结果与实验数据基本吻合。 由于任何原料通过该模型均能转化为反应分子,在反应规律不变的情况下,可适用于任何原料,具有很好的原料适应性。
传统集总动力学模型和分子集总动力学模型均有较好的预测性能, 但传统集总动力学模型所需参数较少,应用更为简便,而分子集总动力学模型对影响因素考虑更周全,其适用性更强。 因此,在建立加氢裂化动力学模型时应考虑多方面的需求, 选择最优的构建方法,以保障效益的最大化。
3 总结与期望
加氢裂化反应动力学模型研究至今已取得了长足的进步。研究初期受分析手段限制,大多数模型依托油品宏观表征信息加以构建, 如简便的关联动力学模型和以馏程划分的简单(传统)集总动力学模型,这些模型由于输入参数较少,其适用性有限。 随着油品分析技术的发展, 复杂集总模型研究受到重视,此类模型集总不再单一针对馏分划分,而是更深入地将石油烃分子以结构或碳数归类, 对不同产品切割方案均能做较好的预测, 并且对于加氢裂化反应过程中氢耗、温升和压降等也能做准确的计算。进入信息时代,计算机技术的进步推动了“分子炼油”的发展, 催生了以分子管理构建加氢裂化动力学模型技术,在分子层面模拟加氢裂化加工反应过程,从而实现工艺优化,为企业获取最大化效益。不同类型动力学模型优缺点及应用场景如表2 所示。

表2 不同类型加氢裂化反应动力学模型对比
至今为止, 加氢裂化传统集总和关联模型较为简单,不能应对复杂的加氢裂化反应过程,研究时应深入挖掘反应机理,尽可能考虑催化剂、反应器构造等影响参数,进一步提高模型的准确性和适用性。连续分子集总动力学模型存在极多的虚拟组分, 构建模型所需实验、工业数据繁多,模拟计算过程复杂,实际应用受限, 后续研究应将模型与实际应用相关联,针对性地选择分子集总,保证模型准确性的同时尽可能减少参数量,提高模型实用性,使其更好地应用于工业实践。 研究人员也应准确地认识不同模型的优缺点,有效结合并高效利用不同模型的优点,开发一种全面的复合动力学模型(如将传统集总模型高实用性与分子集总模型高准确性结合利用),最终实现对加氢裂化反应过程的精准模拟和优化。
猜你喜欢
空气动力学学报(2022年4期)2022-08-23
黑龙江大学自然科学学报(2022年1期)2022-03-29
农村青少年科学探究(2020年5期)2020-08-18
石油炼制与化工(2019年3期)2019-03-15
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
化工管理(2017年21期)2017-03-04
少儿科学周刊·少年版(2015年1期)2015-07-07
浙江大学学报(工学版)(2015年2期)2015-05-30
山东工业技术(2014年19期)2014-08-15
