
COL2A1基因新发突变导致先天性脊柱骨骺发育不良的遗传分析及产前诊断 *
2021-01-29 02:23:54李素萍金玉霞杨静唐萍张卫华周赤燕沈华祥
临床检验杂志 2020年12期
李素萍,金玉霞,杨静,唐萍,张卫华,周赤燕,沈华祥
(嘉兴学院附属妇儿医院&嘉兴市妇幼保健院 a.产前诊断中心,b.超声科,c. 产科,浙江嘉兴 314051)
先天性脊柱骨骺发育不良(spondyloepiphyseal dysplasia congenita,SEDC)是一种罕见常染色体显性遗传病(MIM:183900),发病率约为1/100 000,其特征是身材矮小,畸形骨骺和椎体扁平,早发性关节退行性变,有时还会表现为近视或视网膜变性伴视网膜脱离等[1-2]。患者童年期常出现进展性脊柱弯曲,最终可能会影响呼吸,此外,还存在其他骨骼体征包括扁平脊椎,髋关节畸形导致股骨内旋和畸形足等[3]。先天性SEDC患者之间的临床表型和影像学特征差异明显,通常是由COL2A1基因变异引起。一项研究证实,在663例先证者中发现了COL2A1基因460个突变类型,并证实其与21个明确的遗传病相关[4]。然而,该基因突变热点与表型相关性的研究报道目前仍较为少见。本研究报道了1例身高矮小的SEDC患儿,对其进行临床诊断、家系基因检测和蛋白质功能预测,发现了COL2A1基因变异位点,为该家系遗传咨询及产前诊断提供证据。
1 对象与方法
1.1研究对象 先证者,男,8岁,患儿家属主诉学步时发现姿势异常,运动智力正常。身体发育较同龄人迟缓,具体表现为身高增长慢,四肢粗短。3岁时曾就诊,当时医生怀疑为软骨发育不全,未进行进一步检测,无家族遗传病史,父母表型正常。现因再次妊娠于2019年2月1日就诊于嘉兴市妇幼保健院产前诊断中心。对先证者及父母进一步体检显示,先证者身高101.2 cm,体重17.3 kg,父亲身高172 cm,母亲身高150 cm。先证者身材匀称,甲状腺未触及,四肢脊柱无明显异常。根据主诉及表现,对患儿进行体格检查,采集患儿血液样本进行生化检查(血常规、生长激素与甲状腺功能)和影像学检查。生化检查结果提示,先证者磷元素含量偏低,镁元素含量偏高;血糖、肝功能及甲状腺功能正常。X线摄影考虑骨骺发育不良可能。故而对患儿行遗传学基因检测,并对其父母及胎儿进行了Sanger测序验证以明确病因。依照《国际遗传性骨病分类标准(2010年版)》,根据该患儿的临床特征、影像特点以及分子遗传学的检测结果,该患儿诊断为SEDC。本研究经嘉兴学院附属妇儿医院伦理委员会批准(批准文号:2018010),患者监护人知情同意。
1.2主要仪器及试剂 Agilent 2100 Bioanalyzer (美国安捷伦科技公司),MGISEQ-2000高通量测序仪(深圳华大基因生物医学工程公司),ABI 3730XL测序仪(美国赛默飞世尔科技公司)。MagPure Buffy Coat DNA Midi KF Kit(广州美基生物科技公司),Segmentase随机打断酶(深圳华大基因生物医学工程公司),基因片段捕获探针(美国Roche NimbleGen公司)。
1.3方法
1.3.1基因组DNA提取 采用EDTA-K2抗凝管抽取先证者及其父母外周血各2 mL,另抽取胎儿羊水10 mL,按照MagPure Buffy Coat DNA Midi KF Kit说明书提取DNA,提取样本的质控标准:DNA浓度≥20 ng/μL,总量≥1 000 ng。提取后将样本置于-20 ℃保存。
1.3.2高通量测序 采用华大基因研究院定制的合成芯片,应用目标捕获高通量测序法,对454个基因可能导致的497种与遗传性骨病相关的单基因疾病进行基因检测。步骤:将制备好的DNA片段进行末端补平,接头连接,经Non-Captured PCR构建完整的文库。通过检测芯片上带有生物素的单链DNA探针进行杂交,利用链霉亲和素标记的磁珠将选取的特定基因的外显子(Exon)及侧翼±30 bp区域进行富集,经PCR扩增富集后产物,对PCR产物进行qPCR检测获得序列捕获杂交效率。文库质控标准为子文库浓度≥40 ng/μL,总量≥1 000 ng,子文库片段大小300~550 bp。质控标准:10 ng/μL≤洗脱浓度≤50 ng/μL,洗脱文库片段大小300~550 bp。qPCR检测质控合格后,将一定数量的文库制备DNA纳米球(DNB)后进行上机测序,对数据进行分析质控,信息分析质控标准为去重深度≥100X,30X覆盖度≥95%,Q20≥90%,Q30≥85%,得到原始测序数据。
1.3.3变异致病性分析 对下机的原始数据进行测序质量评估,去除低质量以及被接头污染的reads。采用BWA软件与HG19/HG20进行序列比对,进行序列捕获效果评价,用GATK软件进行单核苷酸变异(SNV)和Indel的查询,生成目标区域碱基多态性结果,进行数据库(NCBI dbSNP, HapMap, 1000 human genome dataset 和 database of 100 Chinese healthy adults)比对,并对找出的可疑突变进行注释、筛选。对检测到的变异,基于HGMD、OMIM数据库、ClinVar数据库以及各大骨病致病突变数据库及文献搜索查阅分析,同时使用 PolyPhen-2(Polymorphism Phenotyping version 2)软件对错义变异进行功能预测和致病性分析,参考美国医学遗传学和基因组学学院(American College of Medical Genetics and Genomics, ACMG)相关指南[5]对变异进行致病性判读。
1.3.4基因突变验证 采用Sanger测序法,使用ABI 3730XL测序仪对先证者检出的变异进行父母和胎儿验证。
2 结果
2.1生化指标及基因检测结果 先证者的生化检测结果见表1。先证者存在COL2A1基因(NM_001844)第50个外显子的c.3589G>A(p.Gly1197Ser)为杂合变异,先证者父母及胎儿均未发现该位点变异(图1)。
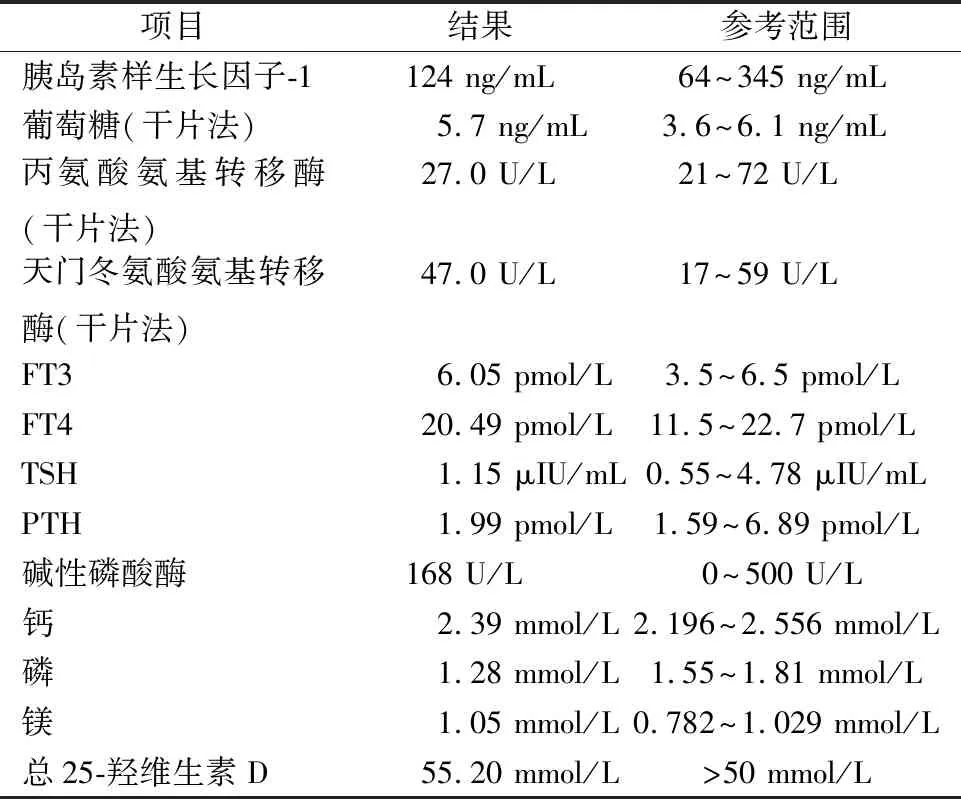
表1 先证者各生化指标检查结果
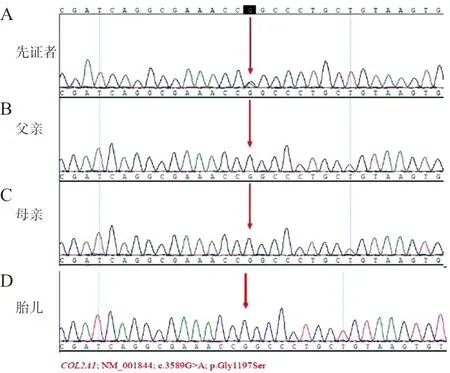
注:A,先证者c.3589G>A(p.Gly1197Ser)位点突变验证结果;B,与先证者突变位点的同一位置的父亲碱基序列;C,与先证者突变位点的同一位置的母亲碱基序列;D,与先证者突变位点的同一位置的胎儿碱基序列。
2.2致病性和保守性分析 采用PolyPhen-2及Mutation Taste软件对COL2A1基因新发变异进行蛋白质功能预测及保守性分析,结果表明,PolyPhen-2软件预测值为0.994,考虑为致病性变异;氨基酸序列保守性为高度保守(图2A)。Mutation Taste分析提示该变异为致病变异,该位点氨基酸序列高度保守(图2B)。因此,COL2A1基因c.3589G>A变异可能是该SEDC患儿的致病原因。

注:A,PolyPhen-2软件对COL2A1基因c.3589G>A(p.Gly1197Ser)变异预测结果为0.994(分数越接近于1,致病性可能越大);B,Mutation Taste软件分析不同物种间氨基酸突变位点保守性。
3 讨论
COL2A1基因编码Ⅱ型前胶原蛋白的α1链,该基因的分子缺陷导致Ⅱ型胶原病[6]。Ⅱ型胶原是一种同源三聚体蛋白,三螺旋中的甘氨酸被取代,极大地损害了同源三聚体的组装和稳定性,在骨成熟和骨重塑中也发挥重要作用[7-8]。这种组织特异性表达导致携带COL2A1致病突变的患者具有广泛表型谱的异质性,出现骨骼发育不良,矮小、眼部异常和听力问题等临床特征[9]。根据现有报道,尚未发现COL2A1基因明显的突变热点,但大多数的COL2A1突变发生在螺旋重复序列区域内。由于Ⅱ型胶原的三螺旋结构是从羧基端向氨基端方向折叠的,因此,有学者认为位于C端的突变与N端突变相比具有更严重的临床表型[10]。此外,有学者在一项29例COL2A1基因突变患者临床研究中发现,该基因新突变在中国人群中可导致骨关节炎伴轻度软骨发育不良和骨骺发育不良多发性近视耳聋[11]。这表明突变的类型、位置和周围的序列及其他未知的遗传和表观遗传因素均可能影响到表型,该基因突变谱和相关的表型尚需进一步研究。
Terhal等[12]对93例SEDC或相关疾病患者的临床、影像学和基因型资料进行回顾性分析,结果发现50%以上的患者做过脊柱侧弯、股骨截骨术或髋关节置换术;56%的患者齿状突发育不良;近视率为45%;37%的患者听力异常。其中,检出2例患者发生甘氨酸至丝氨酸的替换,导致了相对轻微的表型。然而,在702位出现的同样蛋白质转换导致了严重的SEDC(p.Gly702Ser)。甘氨酸至丝氨酸的替换可能导致不同的表型,某些甘氨酸至丝氨酸的替换只会产生较温和的骨骼表型。本研究检出1 197位甘氨酸至丝氨酸的替换是一种非保守性氨基酸取代,影响了含有Gly-X-Y重复序列的三螺旋区的甘氨酸残基,经分析提示该变异可能破坏蛋白质的结构进而影响其功能,在人类基因突变数据库中已经报道了影响G-X-Y基序附近甘氨酸残基的错义变异(G1200S, G1200C)与COL2A1相关的疾病有关,临近位点的突变表明该蛋白质区域的功能非常重要。本例患儿表型相对轻微,表现为腰椎形态不完整,骨骺发育不良。有文献报道了1例携带与本例具有相同位点变异的女孩,其头部相对于躯干和四肢较短,面部平坦,下颌较小,有后下颌畸形,U型腭裂;四肢和脊椎排列整齐,胸部、髂骨短而宽[13]。结合以上研究报道,COL2A1基因c.3589G>A变异可以解释患儿骨骼发育不良表型,但该位点目前病例支持证据较少。然而,本研究仅报道1例家系,骨骼异常除受到与遗传相关的基因调控外,仍可能有其他的综合因素参与其中,该位点突变与表型相关性需更多的病例支持。综上所述,本研究证实COL2A1基因新发突变为该患儿的致病原因,为该家系的遗传咨询和产前诊断提供了依据。
致谢:感谢上海交通大学附属新华医院小儿内分泌科余永国教授对临床诊断的指导和支持,感谢浙江博圣生物技术股份有限公司的技术支持。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
上海计量测试(2022年4期)2022-02-01 07:41:18
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
现代园艺(2017年21期)2018-01-03 06:41:32
人间(2015年11期)2016-01-09 13:12:58
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
邵阳学院学报(自然科学版)(2015年2期)2015-06-05 12:22:39
重庆医学(2015年12期)2015-03-05 05:52:54
