
色谱法测定甜叶菊叶片及其提取物中甜菊糖总苷含量的研究
2021-01-19 06:39:18罗勇为钟晓燕姚满芳吴茜茜唐林波易琳捷张平军
甘蔗糖业 2020年6期
罗勇为,钟晓燕,姚满芳,吴茜茜,唐林波,易琳捷,张平军
(1广东省科学院生物工程研究所,广东广州510316;2广东省绿色制糖工程技术研究中心,广东广州510316;3广西桂林锐德检测认证技术有限公司,广西桂林541004;4桂林三金药业股份有限公司,广西桂林541200;5广西糖业集团有限公司,广西南宁530022)
0 引言
甜叶菊提取物在国际市场上作为一种主要的天然甜味剂,广泛应用于食品、包装食品、桌面甜味剂、饮料(包括能量饮料)、膳食补充剂、烘焙产品、奶制品等行业[1-3]。每年全球该产品的销售额高达几亿美金,市场迅速增长,而且将继续保持强劲的增长趋势。随着消费者对饮料产品和食品的成分含量越来越关注,目前许多消费者也开始避免摄入人工及化学合成甜味剂,减糖仍然是制造商取胜的关键所在[4-5]。而天然来源的甜叶菊和接近零热量的市场定位具有明显的优势。2014年,世界卫生组织建议人们将日常糖摄入量减半,以及公众对一些合成甜味剂的“安全性能”表示担忧,使天然来源甜味剂甜菊糖的需求不断攀升。此外,甜叶菊提取物在软饮料和果汁、冰淇淋以及其他各种产品中的应用日益增加,这归因于甜叶菊提取物的高甜度天然甜味剂特性。到2025年甜菊糖有望占全球甜味剂市场份额的 15%~20%。全球甜菊糖甜味剂市场依据类型分为液体、粉末和药片形式。日益增长的肥胖水平加上对糖尿病和心血管疾病的发病风险的关注促使消费者做出更健康的选择,这有利于创造市场对甜菊糖的需求。由于健康问题,合成或人工甜味剂的天然替代品的需求日益增加,再加上植物来源甜味剂需求的增长等因素,预计将在未来5~10年推动甜菊糖市场的增长[6-10]。
由于粉末甜菊糖的可用性强和使用方便,到2025年该市场份额有望占据整个甜菊糖市场的70%。目前,中国已成为全球最大的甜叶菊生产与出口国,从原料到成品如何更好地满足国内外市场的质量控制要求,是行业经久不息的关注点。本文从工业生产以及便捷快速检测的视角初步研究同时测定甜叶菊叶片及其提取物中甜菊糖总苷的方法,有助于提高甜叶菊提取物生产企业的质量控制水平和产品的市场竞争力,促进甜叶菊产业的发展。
1 材料与仪器
1.1 实验材料与试剂
甜叶菊原料来源于黑龙江、新疆、甘肃的甜叶菊种植基地;甜叶菊提取物来源于桂林莱茵生物科技股份有限公司。
标准品:甜菊苷(购自美国 ChromaDex,含量98.53%)、瑞鲍迪苷 A(购自美国 ChromaDex,含量98.85%);其他7种糖苷标准品:瑞鲍迪苷B、瑞鲍迪苷C、瑞鲍迪苷D、瑞鲍迪苷F、杜克苷A、甜茶苷和甜菊双糖苷(均购自 ChromaDex)。无水甲醇,AR 500 mL/瓶,西陇化工股份有限公司;乙腈为色谱级;磷酸:AR 500 mL/瓶,西陇化工股份有限公司;水为Milli-Q超纯水。
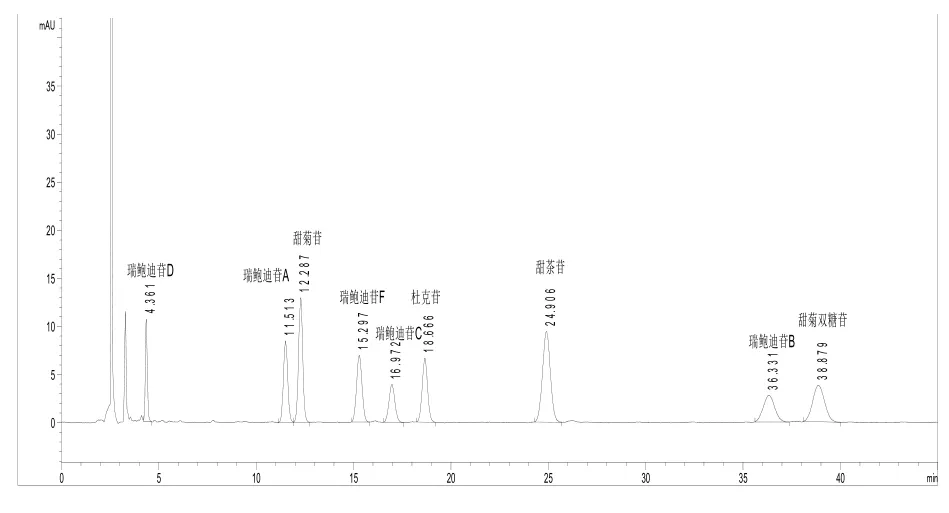
图1 甜菊糖苷9个成分混合标准品图谱
1.2 实验设备与仪器
高效液相色谱仪(型号:1200,美国安捷伦公司),配备在线真空脱气机、四元低压梯度泵、自动进样器、柱温箱、二极管阵列检测器(DAD)及Chemstation色谱工作站;电子分析天平(型号:XS205du,感量:0.01 mg,瑞士梅特勒托利多公司);小型高速离心机(型号:TGL-16B,上海安亭科学仪器厂);超声波清洗器(型号:SK-7200H,上海科导超声仪器有限公司)。
2 实验方法
2.1 色谱条件
以十八烷基键合硅胶填充柱(4.6 mm×250 mm,5 µm)或同类型柱;以乙腈-0.1%磷酸水(30∶70,V/V)为流动相;柱温40℃;流速1.0 mL/min;检测波长为210 nm。理论板数按瑞鲍迪苷A峰计算应不低于3000。
2.2 标准品、样品溶液的制备
2.2.1 混合标准品溶液的制备
分别称取适量甜菊苷、瑞鲍迪苷 A、瑞鲍迪苷B、瑞鲍迪苷C、瑞鲍迪苷D、瑞鲍迪苷F、杜克苷A、甜茶苷、甜菊双糖苷标准品,置于同一个容量瓶中,用30%乙腈水溶液完全溶解后定容,制成每1 mL约含0.04 mg的混合标准品溶液。混合标准品溶液用于确定9种糖苷的相对保留时间。甜菊糖苷9个成分混合标准品图谱如图1所示。
2.2.2 标准品溶液的制备
分别取瑞鲍迪苷 A和甜菊苷标准品适量,加30%乙腈水溶液制成每1 mL含1.0 mg的溶液,即得瑞鲍迪苷A和甜菊苷混合标准品溶液,其图谱见图2。
2.2.3 甜叶菊叶片样品溶液的制备
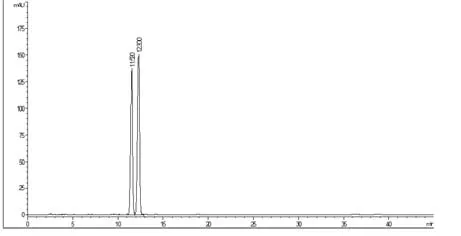
图2 瑞鲍迪苷A和甜菊苷混合标准品图谱
检查甜叶菊原料是否添加甜叶菊梗或其他杂质,进行以下操作。
2.2.3.1 若未含有添加甜叶菊梗或其他杂质
目测无明显的杂质则直接取约50 g原料打粉,用50 mL容量瓶称取样品约2000 mg,加入35~40 mL甲醇浸泡1 h,超声提取30 min,冷却至室温,定容至刻度,摇匀,离心,过0.45 μm滤膜,续滤液转移至进样瓶,待测。
2.2.3.2 若含有添加甜叶菊梗或其他杂质
目测有梗或较多杂质的样品则需取约200 g按四分法分成4份,任取一份原料打粉过100目筛,分别对上、下部分取样,按2.2.3.1步骤进行样品溶液的制备;计算上、下部分的加权含量。过筛部分应超过总打粉量的80%。
2.2.4 甜叶菊提取物样品溶液的制备
取本品粉末约25 mg,精密称定,置25 mL容量瓶中,加30%乙腈水溶液超声处理,冷却至室温,30%乙腈水溶液稀释至刻度,摇匀,滤过,取续滤液,即得样品溶液。其中,甜叶菊原料样品图谱和甜叶菊提取物样品图谱见表3、表4。
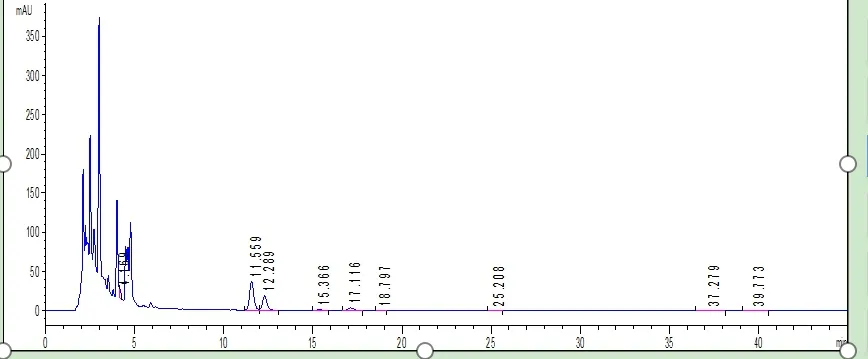
图3 甜叶菊原料样品图谱
2.3.5 测定法
分别精密吸取混合标准品溶液、标准品溶液与样品溶液各 10 µL,注入液相色谱仪,测定。分别对混合标准品溶液、标准品溶液和样品溶液进行色谱分析。将样品溶液的色谱图与混合标准品溶液的色谱图相比较,以确定样品溶液色谱图中各组分对应的峰。记录样品溶液色谱图中甜菊苷、瑞鲍迪苷A、瑞鲍迪苷B、瑞鲍迪苷C、瑞鲍迪苷D、瑞鲍迪苷 F、杜克苷 A、甜茶苷、甜菊双糖苷的峰面积及标准品溶液色谱图中甜菊苷和瑞鲍迪苷 A的峰面积。
瑞鲍迪苷A含量(以干基计)的质量分数wa按公式⑴计算:
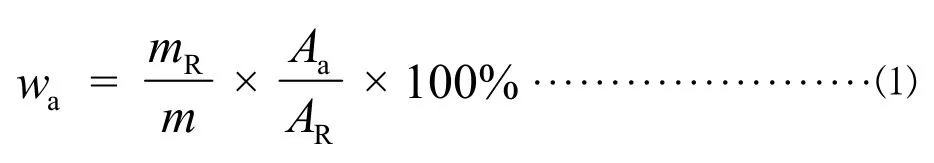
式中:mR-瑞鲍迪苷 A标准溶液中瑞鲍迪苷 A的质量(以干基计),单位为毫克(mg);m-样品溶液中样品的质量(以干基计),单位为毫克(mg);Aa-样品溶液色谱图中瑞鲍迪苷A的峰面积值;AR-瑞鲍迪苷A标准溶液色谱图中瑞鲍迪苷A的峰面积值。
图4 甜叶菊提取物样品图谱
其他8种糖苷含量(以干基计)的质量分数wi按公式⑵计算:

式中:i-s、b、c、d、f、da、ru、sb,分别对应甜菊苷、瑞鲍迪苷 B、瑞鲍迪苷 C、瑞鲍迪苷 D、瑞鲍迪苷F、杜克苷A、甜茶苷、甜菊双糖苷;mS-甜菊苷标准溶液中甜菊苷的质量(以干基计),单位为毫克(mg);m-样品溶液中样品的质量(以干基计),单位为毫克(mg);fi-i组分与甜菊苷的式量比值:1.00(甜菊苷)、1.00(瑞鲍迪苷 B)、1.18(瑞鲍迪苷 C)、1.40(瑞鲍迪苷 D)、1.16(瑞鲍迪苷 F)、0.98(杜克苷A)、0.80(甜茶苷)、0.80(甜菊双糖苷);Ai-样品溶液色谱图中i组分的峰面积值;AS-甜菊苷标准溶液色谱图中甜菊苷的峰面积值。
由上述公式⑴和⑵计算得到 9种组分的含量wa、ws、wb、wc、wd、wf、wda、wru和wsb,取各组分含量之和即为样品中甜菊糖苷含量。
3 实验结果与讨论
3.1 色谱系统稳定性试验
取甜叶菊瑞鲍迪苷A溶液,连续进样6针;①重复性:6次测试值的峰面积及保留时间的RSD值,以瑞鲍迪苷A计,峰面积的RSD值应不高于1.0%,保留时间的RSD值应不高于2.0%;②以瑞鲍迪苷A对称因子计,应在0.8~1.5之间;③分离度应不低于 1.5;④理论塔板数,以瑞鲍迪苷 A理论塔板数计,应不低于3000。结果如表1、表2所示。
结果表明:瑞鲍迪苷A的保留时间为11.5 min左右,峰面积的RSD 0.082%,远低于1.0%,保留时间的RSD 0.35%,低于2.0%,重复性良好;瑞鲍迪苷A对称因子1.13,在0.8~1.5之间;瑞鲍迪苷A的分离度1.86,高于1.5,主成分与相邻杂质峰分离良好;瑞鲍迪苷 A理论塔板数 13803,远高于3000,柱效较高;整体表明检测方法采用的色谱系统稳定性良好。
3.2 线性分析
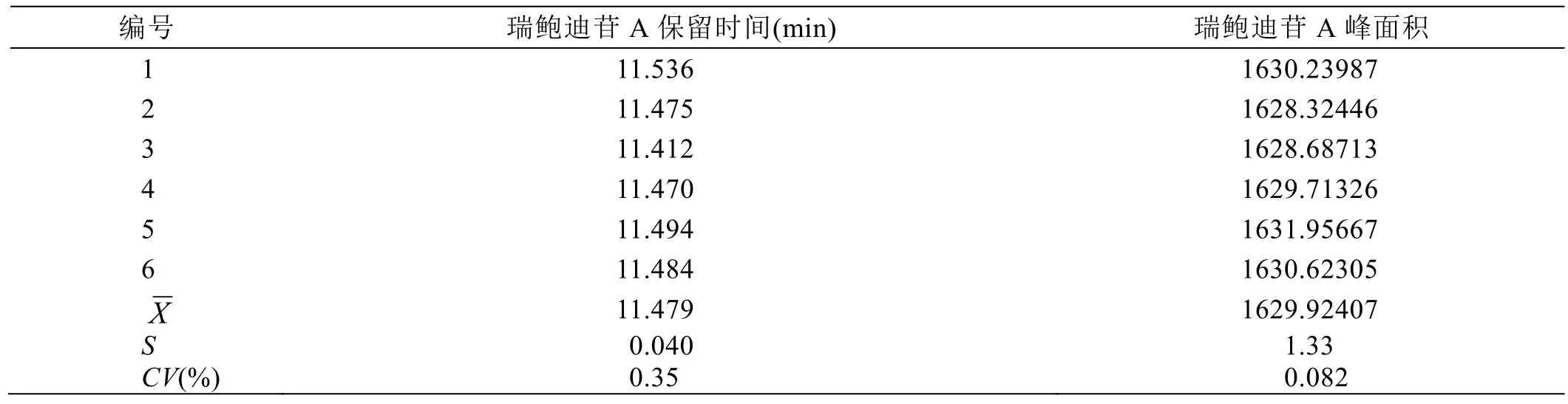
表1 重复性试验结果

表2 色谱系统稳定性试验结果
瑞鲍迪苷A、甜菊糖苷标准品溶液曲线的制备:精密称取瑞鲍迪苷A 49.43 mg,甜菊糖苷50.23 mg,分别置于25 mL容量瓶中,加入乙腈水溶液超声溶解,放置至室温,乙腈水定容至刻度,摇匀后做储备液使用。用移液管移取适量,乙腈水定容至刻度,依次逐级稀释制备其他5个不同浓度的溶液,将以上制备好的6个不同的标准品溶液各10 μL分别注入HPLC仪,制备瑞鲍迪苷A、甜菊糖苷标准溶液曲线。结果如表3、表4所示。
结果表明:瑞鲍迪苷 A线性回归方程:Y=1943.9444X+1.4026,相关系数R2=0.999994;甜菊糖苷线性回归方程:Y=2330.3740X+3.7848,相关系数R2=0.999990;在0.001~2.0 mg/mL浓度范围内与瑞鲍迪苷A、甜菊糖苷峰面积呈良好线性关系。
3.3 稳定性分析
取甜叶菊提取物溶液,间隔一定时间进样1次,记录峰面积,结果见表5。
试验结果表明,不同时间同一甜叶菊提取物溶液进样,瑞鲍迪苷 A峰面积的相对标准偏差为0.12%,甜菊糖苷峰面积的相对标准偏差为0.18%,均小于2%,甜叶菊样品溶液在24 h内稳定。

表3 甜叶菊瑞鲍迪苷A曲线的制备
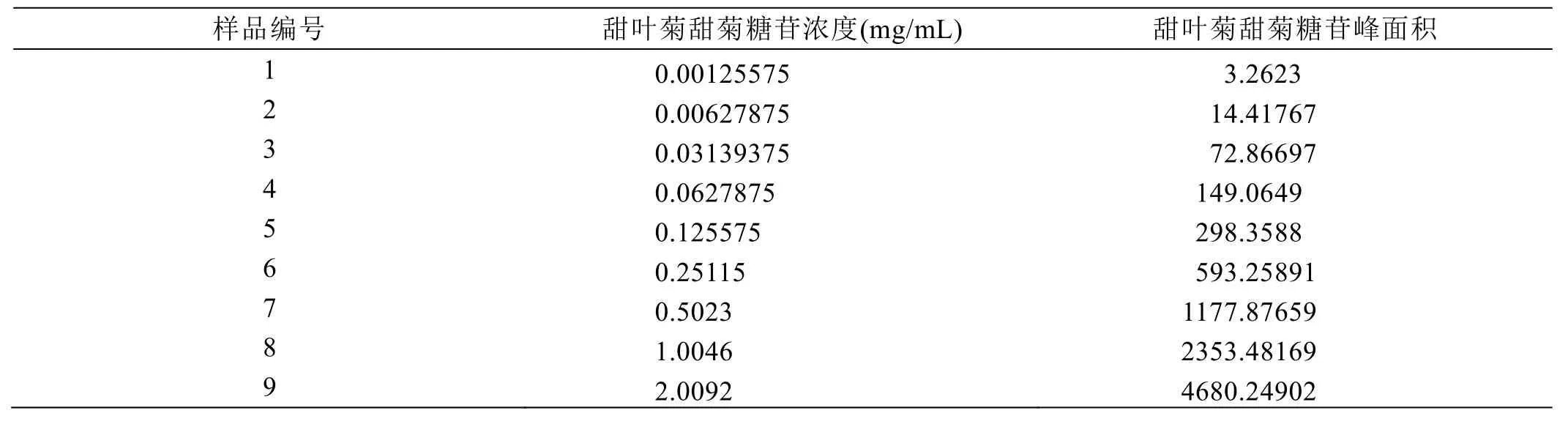
表4 甜叶菊甜菊糖苷曲线的制备
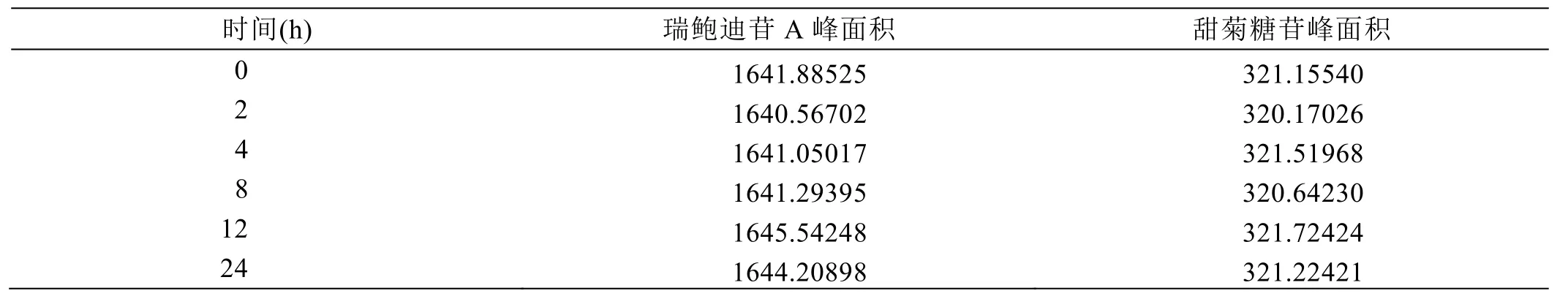
表5 甜叶菊提取物溶液稳定性考察
3.4 准确度验证
精密称取甜叶菊提取物9份,以3份为一个浓度,分别加入标准溶液为甜叶菊提取物中瑞鲍迪苷A、甜菊糖苷含量的40%、90%、130%,再加入30%乙腈水超声溶解,冷却至室温,30%乙腈水定容至刻度,摇匀,0.45 μm微孔滤膜滤过,取续滤液进行HPLC分析,回收率结果见表6、表7。
结果表明,瑞鲍迪苷A加样回收的平均回收率为100.32%,相对标准偏差0.47%;甜菊糖苷加样回收的平均回收率为99.66%,相对标准偏差0.47%;回收率结果均在95%~105%之间,方法有良好的准确度。
3.5 耐用性分析
精密称取甜叶菊提取物24.88 mg,置于25 mL容量瓶中,加入30%乙腈水超声溶解,冷却至室温,30%乙腈水定容至刻度,摇匀,0.45 μm微孔滤膜滤过,取续滤液进行HPLC分析。取不同品牌的色谱柱、更改色谱柱的温度、更改流动相流速分别进行样品含量的测定,测试结果见表8。
试验结果表明,采用不同品牌的色谱柱,瑞鲍迪苷A含量的相对标准偏差为0.32%,甜菊糖苷峰含量的相对标准偏差为0.85%;更改色谱柱的温度,瑞鲍迪苷A含量的相对标准偏差为0.19%,甜菊糖苷峰含量的相对标准偏差为 0.36%;改变流动相的流速,瑞鲍迪苷A含量的相对标准偏差为0.21%,甜菊糖苷峰含量的相对标准偏差为 0.28%;上述含量RSD均小于2%,说明本检测方法在测定条件有小的变动时,测定结果不受影响,具有较好的耐用性。由试验可知,当采用绿百草-C18,1.0 mL/min的流速,色谱柱温度40℃时目标峰峰型尖锐,主成分与相邻杂质峰分离良好,柱效较高,条件最优。
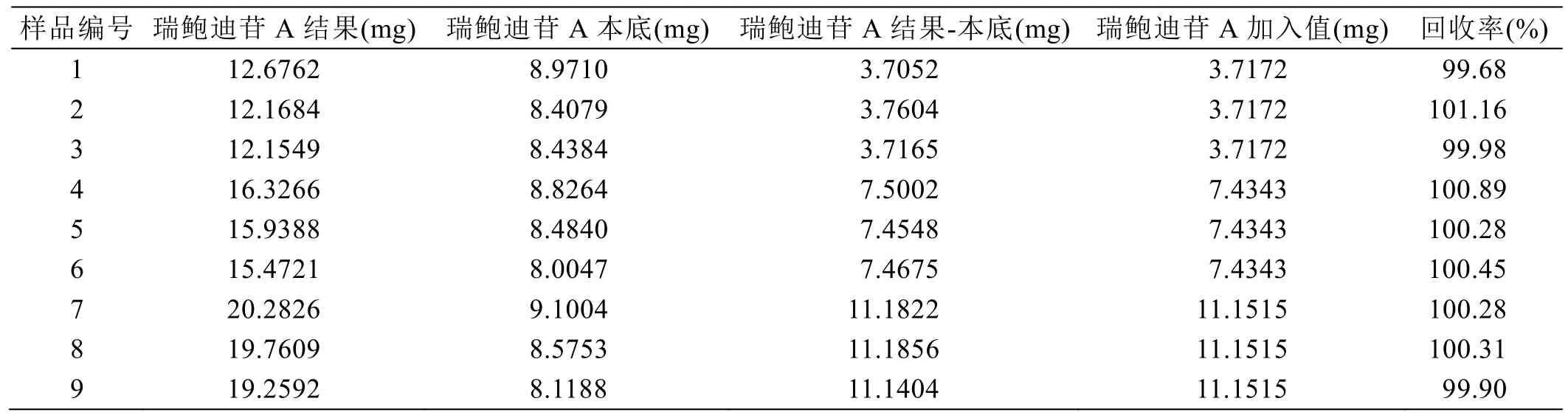
表6 甜叶菊瑞鲍迪苷A回收率试验结果
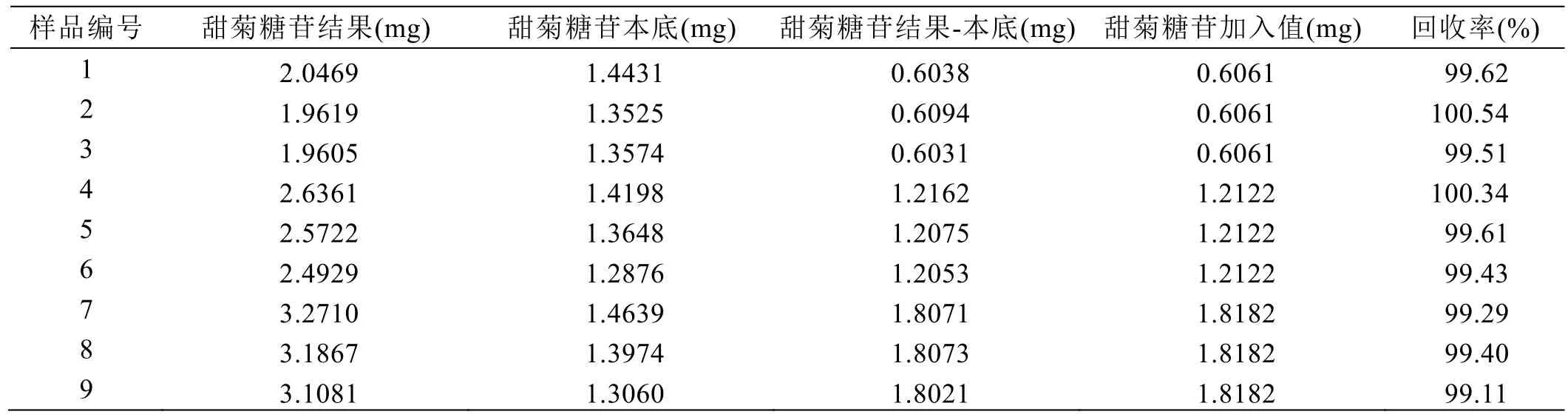
表7 甜叶菊甜菊糖苷回收率试验结果
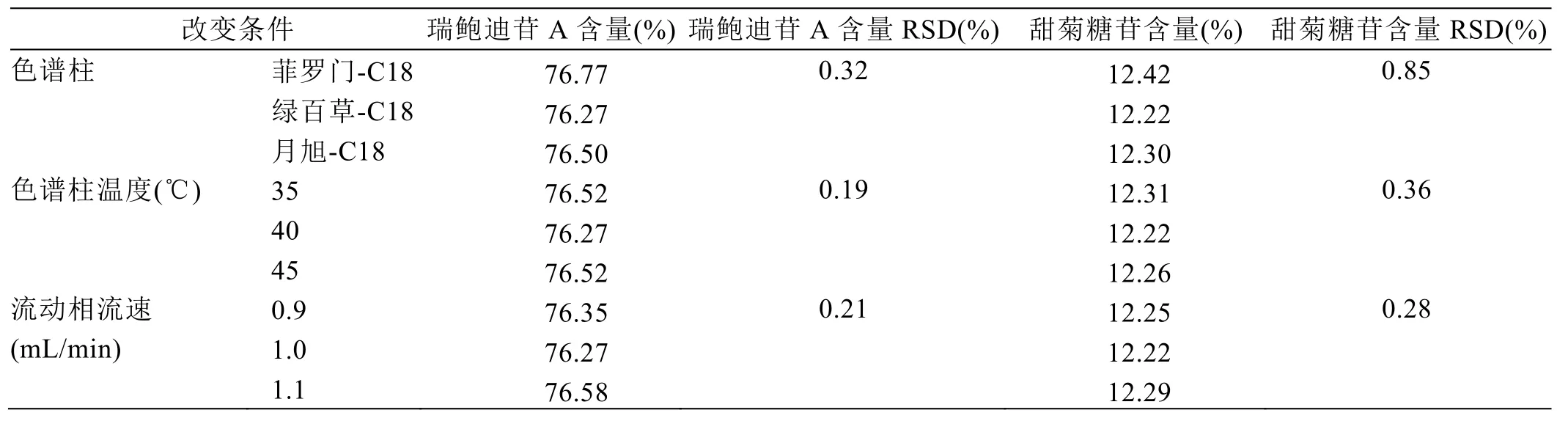
表8 耐用性试验结果汇总
3.6 甜叶菊原料杂质的影响
基于行业现状,市场上销售的甜叶菊原料大多含有不同程度的小石子、土、甜叶菊梗茎等杂质,为了避免因含杂质太高原料品质变差而导致需要改进工艺,从而提高生产成本,业内提出双12%标准:杂质占比不超过12%,原料总苷含量不低于12%。为了提高检测甜叶菊原料的效率并避免杂质对结果的影响,取不含杂质的甜叶菊加入不同的杂质量进行测试。试验结果见表9。
结果表明,不同的杂质添加量与过筛部分含量呈正相关,当过100目筛部占总打粉重量的80%时,较为接近业内的双12%标准。所以过100目筛部分的甜叶菊原料应超过总打粉重量的 80%;同时,对于杂质较多的样品,为了消除杂质对甜叶菊含量的干扰,可通过对 100目筛上(通过 100目筛样品)、筛下(未通过 100目筛样品)部分的样品粉末分别检测,计算加权含量。
4 讨论与结论
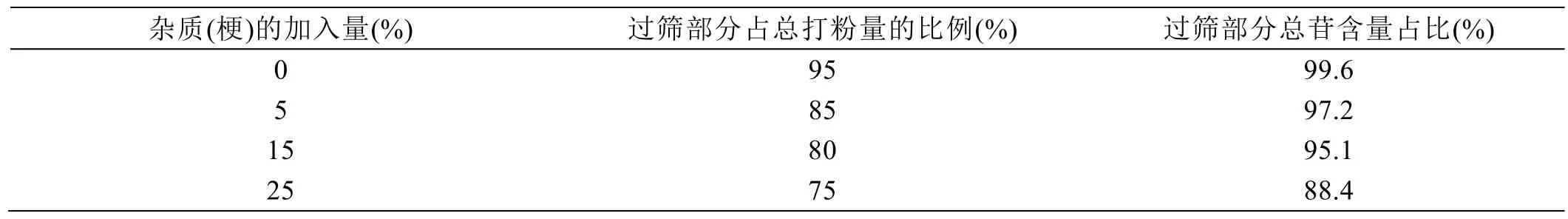
表9 甜叶菊原料中杂质与过筛部分总苷的关系
本方法采用甜叶菊原料打粉过100目筛部分的 应超过总打粉重量的 80%,通过计算上、下部分的加权含量能确保消除杂质对原料的干扰,大大缩短甜叶菊原料的制样时间的同时保证结果的准确性。色谱条件选择绿百草-C18,乙腈:0.1%磷酸水洗脱,流动相无需使用缓冲盐体系,任一实验室均简便易配且能延长色谱柱的使用寿命,有着良好的分离度和较高的柱效,色谱柱的分离度和使用寿命优于甜菊糖苷国标方法和文献所述氨基柱方法[11]。加样回收的瑞鲍迪苷 A和甜菊苷的平均回收率分别为99.66%、100.32%;相对标准偏差0.47%,满足食品理化检测中检测方法确认的技术要求中关于回收率参考范围95%~105%的规定,表明该法有良好的准确度,符合方法验证要求。校准曲线浓度范围覆盖3个数量级,相关系数不低于 0.99,表明该法具有较好的线性范围,符合方法验证要求。本方法的重现性、稳定性相对标准偏差均小于2%,满足方法验证的要求。本方法在测定条件有小的变动时,测定结果不受影响,具有较好的耐用性。本方法目标峰能与相邻的杂质有效分离,分离度及纯度因子满足要求,具有良好的专属性。
甜菊糖苷RP-HPLC分析方法的验证结果表明,该分析方法有良好的精密度和较高的准确度,线性范围符合要求,能够准确、快速、有效地检测出甜叶菊叶片及提取物中甜菊糖总苷的含量,控制各环节产品的质量。
猜你喜欢
食品工业(2021年5期)2021-06-10 06:47:00
艺术品鉴(2020年6期)2020-12-06 10:49:08
天然产物研究与开发(2018年6期)2018-07-09 06:01:50
领导文萃(2017年6期)2017-03-24 09:31:39
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
中国糖料(2016年1期)2016-12-01 06:49:01
天津科技大学学报(2016年1期)2016-02-28 16:59:44
食品界(2016年4期)2016-02-27 07:36:47
生物技术通报(2015年9期)2015-10-25 07:02:21
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
